E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 3(2); 2010 > Article
-
Case Report
A Case of Juvenile Huntington Disease in a 6-Year-Old Boy - Jun-Sang Sunwoo, Soon-Tae Lee, Manho Kim
-
Journal of Movement Disorders 2010;3(2):45-47.
DOI: https://doi.org/10.14802/jmd.10012
Published online: October 30, 2010
Department of Neurology, Seoul National University Hospital, Seoul, Korea
- Corresponding author: Manho Kim, MD, PhD, Department of Neurology, Seoul National University Hospital, 101 Daehak-ro, Jongno-gu, Seoul 110-744, Korea, Tel +82-2-2072-2193, Fax +82-2-3672-7553, E-mail kimmanho@snu.ac.kr
• Received: September 6, 2010 • Accepted: September 25, 2010
Copyright © 2010 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 9,215 Views
- 65 Download
- 4 Crossref
ABSTRACT
- Huntington disease is a neurodegenerative disorder distinguished by the triad of dominant inheritance, choreoathetosis and dementia, usually with onset in the fourth and fifth decades. It is caused by an unstable cytosine-adenine-guanine (CAG) trinucleotide repeat expansion in the gene IT15 in locus 4p16.3. Juvenile HD that constitutes about 3% to 10% of all patients is clinically different from adult-onset form and characterized by a larger number of CAG repeats typically exceeding 60. We report a case of a 6-year-old boy with myoclonic seizure and 140 CAG repeats confirmed by molecular genetic analysis.
- The patient was a 6-year-old boy with normal birth. His father was previously diagnosed with HD and presented with choreoathetoid movement at the age of 32 years. Molecular genetic analysis confirmed the CAG repeats expansion ([CAG]55/[CAG]18). For the purpose of genetic counseling, the patient had presymptomatic genetic test at the first 6 months of life. Polymerase chain reaction (PCR)-based analysis demonstrated 140 CAG repeats in one allele ([CAG]140/[CAG]20), much greater than that of his father.
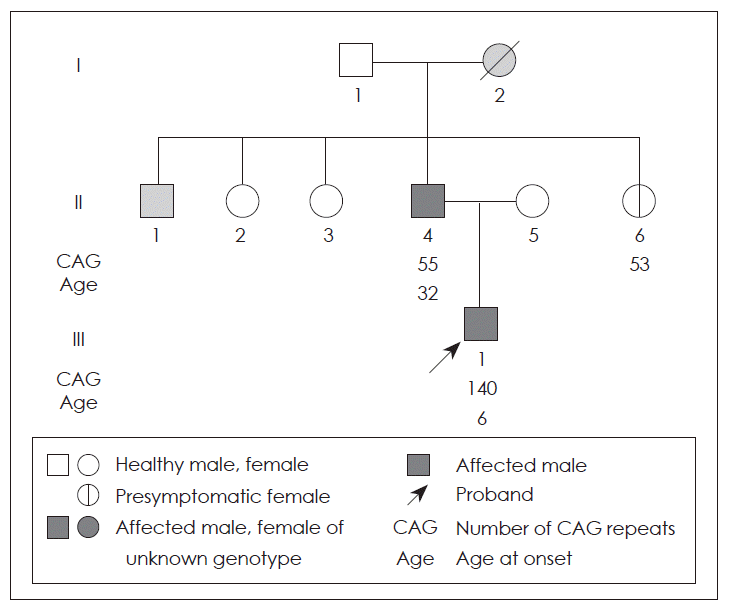
- Family history revealed that patient’s paternal grandmother had similar kind of abnormal movements and died at 50 years of age. The patient’s father had four siblings (one brother and three sisters) and the elder brother has been bedridden since a few years ago with a history of abnormal movements. All his sisters were healthy and presymptomatic genetic examination disclosed that the repeat sequence of the younger sister was expanded with repeat length 53 and those of others were normal. The family pedigree is shown in Figure 1.
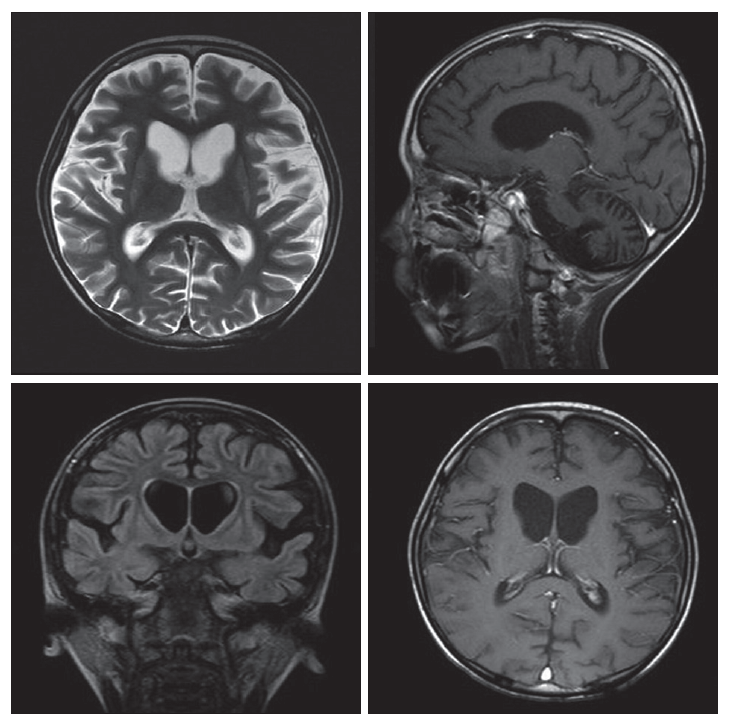
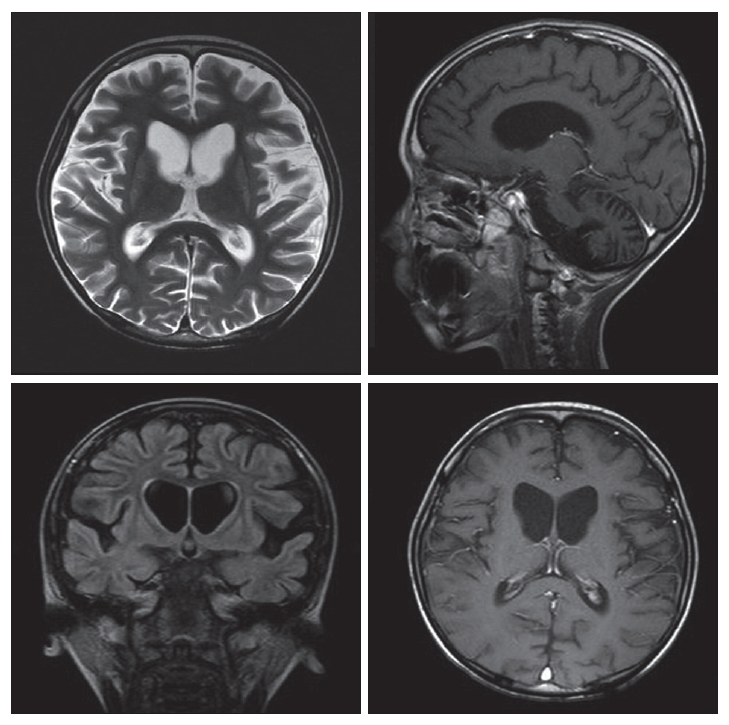
- At the age of 6 years, the patient presented to this hospital because of myoclonic seizure. His weight was 13 kg and developmental regression was observed. An electroencephalogram showed frequent generalized sharp wave and spike discharges. Magnetic resonance imaging (MRI) revealed atrophy of the bilateral putamen, caudate nucleus and hemispheric cerebellum with enlargement of ventricles (Figure 2). Treatment with valproate was begun, and episodes of absence and generalized tonic-clonic seizure as well as myoclonic seizure persisted approximately 3 times per month. At 7 years, he visited the emergency room due to fever and recurrent seizure. Seizure activity was improved after administration of lorazepam. We observed dystonia, choreic movement and spasticity in all limbs. He underwent percutaneous endoscopic gastrostomy because of swallowing difficulty. Further course of the disease was characterized by gradual aggravation of spasticity and dystonic posture, and he could not walk any more since age 8.
Case
- HD is a progressive neurodegenerative disorder of which George Huntington made the first description in 1872. The prevalence of HD in the West is estimated at 5 to 10 persons per 100,000 population, and the prevalence in Asia is approximately one tenth of the Western population. Juvenile HD with an age of onset before 20 years occurs less frequently than usual adult-onset HD, and only 1% to 2% of patients develop signs in the first decade of life. Juvenile HD has been predominantly in single case reports, and to our knowledge only one case of juvenile HD with genetic confirmation was reported in Korea.3
- The disease is a trinucleotide repeat disorder which is caused by an abnormal CAG repeat expansion in the gene IT15. This mutation results in synthesis of a modified protein known as huntingtin with increased polyglutamine domain. The number of repeats is usually below 30 in the normal population and the pathogenic number of repeats is more than 40 in the patients. The length of repeat sequence detected by molecular genetic analysis determined the age of onset as well as the presence of disease. There is negative correlation between the age at onset and the repeat length, and early-onset form with onset before 10 years have a higher number of repeats exceeding 80.4 Early age of onset is also related to paternal inheritance as a result of high instability of CAG repeats during spermatogenesis. Likewise, this patient revealed paternal transmission with 140 CAG repeats, which is much larger than 92 repeats of previously reported case in Korea.3
- Adult HD is characterized clinically by a triad of choreic movement, neuropsychiatric manifestation and cognitive disturbance. These symptoms also can be observed in older adolescents, while the children with the early age of onset may have a more distinct clinical presentation. The most common presenting symptoms of juvenile HD in the first decade of life are declining cognitive function, followed by behavioral disturbance, rigidity, oropharyngeal dysfunction and seizures.5 Seizure, the first symptom of this patient, is a common feature of juvenile HD. It was reported that epilepsy occurred in more than 80% of patients with onset in the first 10 years.6 Generalized tonic-clonic, myoclonic and absence seizures are common seizure types. Epileptiform discharges include generalized polyspike and slow-wave complex, spike and slow-wave complex, multifocal spikes and focal spikes. In agreement with previous study, additional symptoms and signs that developed during the course of the disease were dystonia, spasticity, chorea and dysphagia.7 Chorea is very uncommon as a presenting symptom in juvenile HD, but may occur during the course of the disease. The progression of disease in this patient showed a rapid downhill course. Two years after the first manifestation, the patient became wheelchair-bound. In general the younger age at onset is related to more rapid progression of disease and shorter survival of patients.7
- We report characteristic clinical, laboratory and radiological findings of juvenile HD in a 6-year-old boy. The diagnosis was confirmed by PCR-based genetic analysis which detected 140 CAG repeats in a pathogenic allele. To our knowledge, it is the largest number of repeats in the cases of juvenile HD reported in Korea. It is important to identify that juvenile HD can present with variable clinical phenotypes, which are different from adult-onset HD.
Discussion
- This work was supported by grants from the Korea Health 21 R&D Project (A092058), FPR08K1301-02210 and WCU Neurocytomics.
Acknowledgments
Acknowledgments
Figure 1Pedigree of the patient. I-2 had a history of involuntary movement and deceased at 50 years of age. II-1 had similar kind of abnormal movement and had been bed-ridden state since a few years ago. But genetic test of HD was not performed for both of them. II-4, the patient’s father, was diagnosed of HD at 32 years. II-6 and III-1(the patient) was diagnosed at the age of 30 years and 6 months respectively by presymptomatic test. HD: Huntington disease.

Figure 2MRI of the patient. Brain MRI showed atrophic change of putamen, caudate nucleus, cerebral cortex and cerebellum. Flattening of wall of frontal horn with ventriculomegaly is remarkable. There is no abnormally enhancing lesion. MRI: magnetic resonance image.
- 1. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993;72:971–983.ArticlePubMed
- 2. Nance MA, Myers RH. Juvenile onset Huntington’s disease--clinical and research perspectives. Ment Retard Dev Disabil Res Rev 2001;7:153–157.ArticlePubMed
- 3. Lee MS, Shin CH, Son DW, Park KH, Kim DH, Kim KY, et al. A case of juvenile Huntington’s disease confirmed by molecular genetic analysis. J Korean Child Neurol Soc 1999;7:113–118.
- 4. Wojaczyńska-Stanek K, Adamek D, Marszał E, Hoffman-Zacharska D. Huntington disease in a 9-year-old boy: clinical course and neuropathologic examination. J Child Neurol 2006;21:1068–1073.ArticlePubMed
- 5. Nance MA. Genetic testing of children at risk for Huntington’s disease. US Huntington Disease Genetic Testing Group. Neurology 1997;49:1048–1053.ArticlePubMed
- 6. Landau ME, Cannard KR. EEG characteristics in juvenile Huntington’s disease: a case report and review of the literature. Epileptic Disord 2003;5:145–148.ArticlePubMed
- 7. Gonzalez-Alegre P, Afifi AK. Clinical characteristics of childhood-onset (juvenile) Huntington disease: report of 12 patients and review of the literature. J Child Neurol 2006;21:223–229.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
Citations to this article as recorded by 
- Drug-Resistant Epilepsy in Children with Juvenile Huntington's Disease: A Challenging Case and Brief Review
Abdulhafeez M. Khair MD, Jessica Kabrt DO, Stephen Falchek MD
Qatar Medical Journal .2020;[Epub] CrossRef - Introducing an expanded CAG tract into the huntingtin gene causes a wide spectrum of ultrastructural defects in cultured human cells
Ksenia N. Morozova, Lyubov A. Suldina, Tuyana B. Malankhanova, Elena V. Grigor’eva, Suren M. Zakian, Elena Kiseleva, Anastasia A. Malakhova, Hiroyoshi Ariga
PLOS ONE.2018; 13(10): e0204735. CrossRef - Tics as an initial manifestation of juvenile Huntington’s disease: case report and literature review
Shi-Shuang Cui, Ru-Jing Ren, Ying Wang, Gang Wang, Sheng-Di Chen
BMC Neurology.2017;[Epub] CrossRef - Neuropathological Comparison of Adult Onset and Juvenile Huntington’s Disease with Cerebellar Atrophy: A Report of a Father and Son
Caitlin S. Latimer, Margaret E. Flanagan, Patrick J. Cimino, Suman Jayadev, Marie Davis, Zachary S. Hoffer, Thomas J. Montine, Luis F. Gonzalez-Cuyar, Thomas D. Bird, C. Dirk Keene
Journal of Huntington's Disease.2017; 6(4): 337. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite