E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 5(2); 2012 > Article
-
Case Report
A Case of Multiple System Atrophy-Cerebellar Type Preceded by Dementia - Eun Hye Jang, Joo Kyung Lee, Hyun Jung Jang, Mi-Jung Kim, Sun Ju Chung
-
Journal of Movement Disorders 2012;5(2):48-52.
DOI: https://doi.org/10.14802/jmd.12011
Published online: October 30, 2012
Department of Neurology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
- Corresponding author: Sun Ju Chung, MD, PhD, Department of Neurology, Asan Medical Center, University of Ulsan College of Medicine, 88 Olympic-ro 43-gil, Songpa-gu, Seoul 138-736, Korea, Tel +82-2-3010-3440, Fax +82-2-474-4691, E-mail sjchung@amc.seoul.kr
- The authors have no financial conflicts of interest.
• Received: July 20, 2012 • Revised: September 10, 2012 • Accepted: September 10, 2012
Copyright © 2012 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 16,560 Views
- 92 Download
- 3 Crossref
ABSTRACT
- Multiple system atrophy (MSA) is a sporadic, adult-onset disease characterized by progressive degeneration of nervous systems including cerebellar, pyramidal, extrapyramidal, and autonomic system. Although a few recent studies reported that cognitive impairments could occur in patients with MSA, prominent dementia with progressive decline is not a typical clinical manifestation of MSA. In particular, dementia with MSA-cerebellar type is very rare. We have experienced a patient with 2-year history of severe cognitive impairment, who was finally diagnosed as MSA-cerebellar type.
- A 53 years-old male visited to our clinic because of cognitive impairment for two years ago. The patient graduated from university, had worked as an office worker and recently retired. His wife described that, the patient’s memory and ability of judgment had been impaired for 2 years, and his verbal output had been decreased. The patient was not taking any medicines previously. Because of forgetting his medication schedule, he attached his medicine on his calendar. Sometimes he forgot to turn off a gas stove. Although he had never lost his ways, he had trouble with driving and made some car accidents because of misperception. There were also some changes in his behavior. He had turned much introversive instead of sociable previously, and showed indifference towards household matters or works related with their offspring. Inappropriate laughing was observed in serious situations. Without a caregiver, he could not care of his own hygiene or eating. However, there were no psychiatric symptoms such as hallucination or delusion.
- On the first visit, there was no remarkable finding in examination of motor and cerebellar function. A brain magnetic resonance imaging (MRI) showed no remarkable finding. A detailed neuropsychiatric evaluation (Seoul Neuropsychological Screening Battery) was performed. The results were interpreted using 1.0 standard deviation (16 percentile) of normal Korean population aged 55, as a cut-off value. He showed time disorientation, and impairments in encoding, retention and retrieval of verbal memory, and retrieval of visual memory. The frontal lobe functions including executive functions were deteriorated. Considering the progressive decline in intellectual function affecting memory and other cognitive domain, and interference of activities of daily, he was diagnosed with dementia. At the time, the patient was regarded as early frontotemporal dementia. Therefore, we decided to follow up treatment without any medication.
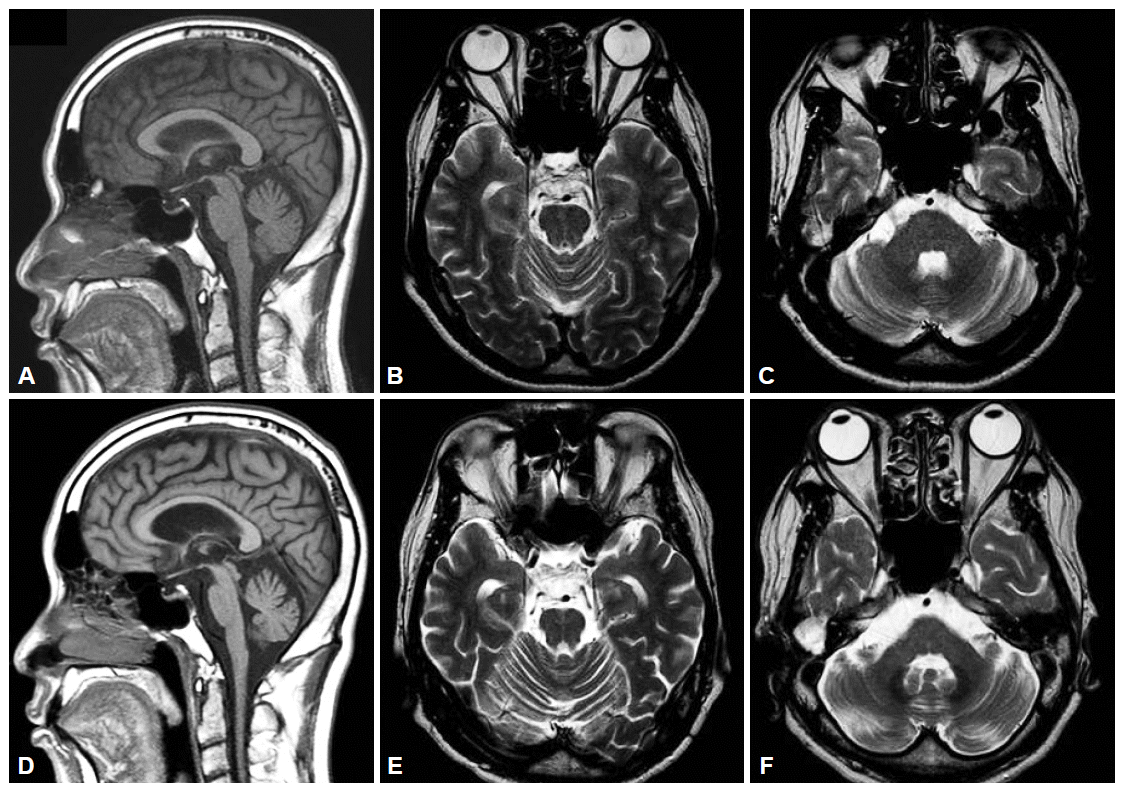
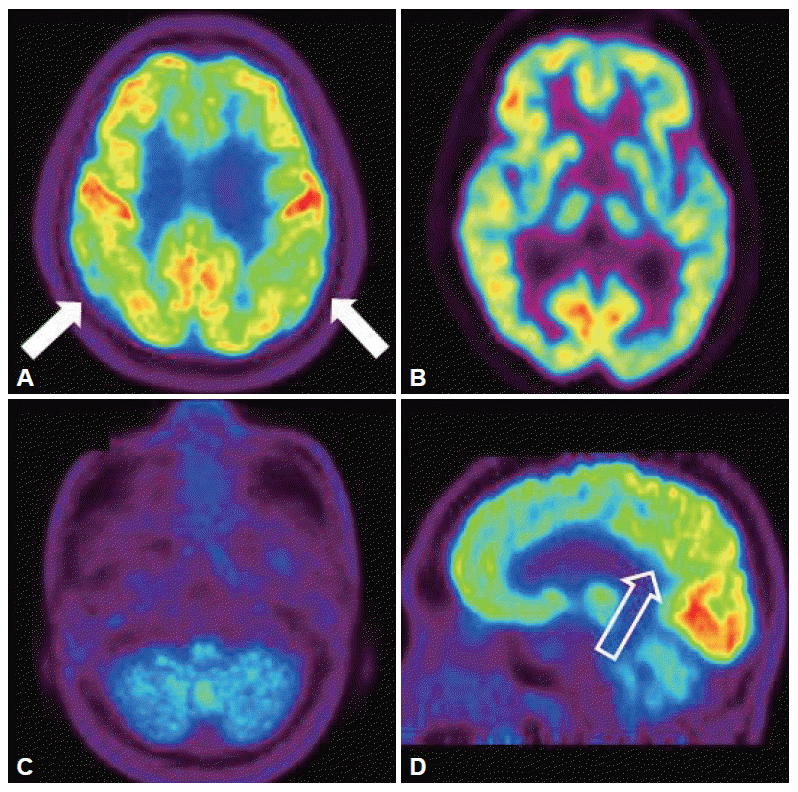
- Six months later, he complained about gait disturbance with a feeling of right leg dragging. In addition, he had suffered from the postural imbalance, and difficulties in swallowing and articulation. He developed the orthostatic dizziness and urinary difficulties, including frequency, nocturia and residual urine sense, had also developed. On neurological examination, he was alert. Cranial nerve examination revealed moderate dysarthria and mild dysphagia. There was no limitation of ocular movements. There were no abnormalities on tests for optokinetic nystamgus or smooth pursuit eye movement. However, hypermetric saccade in eye movement was observed. Motor power was normal. Resting and postural tremor, rigidity, and bradykinesia were not observed. The sensation of all modalities was normal. Deep tendon reflexes were increased in all extremities, Hoffman’s signs were positive, bilaterally. Babinski’s signs were negative. Limb ataxias were observed in both lower extremities. He was unable to tandem gait. The serological tests for syphilis and viruses including HIV were negative. Gene tests for spinocerebellar ataxia type 1, 2, 3, 6, 7, and 17, and dentatorubropallidoluysian atrophy were all negative. The head up tilt test revealed orthostatic hypotension; systolic blood pressure was decreased 31 mm Hg 3 minutes after standing. The volume of residual urine immediately after urination was 110 mL. The neuropsychological test was followed up to evaluate the progression of cognitive dysfunction. Verbal and visual memories and frontal lobe functions, including inhibitory function and semantic word fluency, were significantly deteriorated compared with previous test 6 months ago (Table 1). The results of neuro-psychological testing suggested the possibility of frontotemporal dementia. The follow-up brain MRI has showed newly developed atrophy of cerebellum, pons and middle cerebellar peduncle (Figure 1). Positron emission tomography (PET) was performed using 18F-fluorodeoxyglucose (FDG), and showed decreased cerebral glucose metabolism on the bilateral bagal ganglia, cerebellum, both parietal lobes, and left posterior cingulate gyrus in visual analysis (Figure 2). Finally, based on the features of cerebellar ataxia, autonomic dysfunction and pyramidal dysfunction, he was diagnosed with MSA-cerebellar type (MSA-C).2 He was prescribed with rivastig-mine for dementia and other medications for urinary problems.
Case
- This patient had shown rapid progressions of memory impairment, behavioral changes and impairment in executive functions, and been diagnosed with dementia initially. Thereafter, cerebellar ataxia with dysautonomia and pyramidal dysfunction had occurred, and finally he was diagnosed with MSA-C.
- MSA is a neuro-degenerative disease, which usually occurs sporadically in adults, older than 30 years.2 Typically it is presented with parkinsonism, cerebellar ataxia, dysautonomia and pyramidal dysfunction. Pathologically, neuronal cell loss, gliosis and αSyn-immunopositive GCI are observed in structures of striato-nigral pathway and olivo-ponto-cerebellar pathway. According to the second consensus statement on the diagnosis of MSA,2 MSA can be clinically diagnosed as “probable MSA” or “possible MSA” by their certainty of diagnosis, and as “MSA with predominant parkinsonism; MSA-P” or “MSA with predominant cerebellar ataxia; MSA-C” by the major symptom. However, some findings of so-called “red flags” which do not support MSA are also described. These red flags include dementia on Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition. Therefore, the presence of dementia can exclude the diagnosis of MSA according to these criteria. However, some recent studies have reported 20–40% prevalence of cognitive impairment in MSA.3
- The cognitive impairment in MSA is usually characterized by predominant impairment of frontal lobe functions, such as executive functions, than memory impairment.1,4 Patients with MSA-P showed more severe and diffuse cognitive impairments than patients with MSA-C.5 Comparing MSA with other α-synucleinopathies,6 the severity of cognitive decline was more than PD, but milder than DLB. Compared to patients with DLB, patients with MSA have a lesser impairment in the memory function. However, the visuospatial function and executive function were more damaged in patients with MSA. The frequency of cognitive decline did not show a correlation with disease duration. Patients with MSA and dementia demonstrated frontal lobe atrophy in MRI and decreased perfusion in the frontal and posterior parietal lobes in single photon emission computed tomography.3,4 With regard to temporal relationship of onset between motor symptoms and cognitive impairment in MSA with dementia, patients with MSA preceding dementia showed Alzheimer’s disease (AD)-like pattern of cognitive impairment, severe memory deficits and more confusion than MSA preceding motor symptoms.3 Moreover, imaging studies revealed more diffuse cortical atrophy and severe white matter changes in patients with MSA preceding dementia. Unlike the previous studies, our patient showed severe cognitive impairments in MSA-C and the pattern of cognitive deficit was different from AD despite of preceding dementia.
- The pathogenic mechanisms of cognitive impairment, especially frontal lobe dysfunction in MSA are unclear. The clinical presentation and the frequency of cognitive impairment in MSA may differ from that of other synucleinopathies such as DLB and PD with dememtia (PDD), so it is possible to presume that the mechanisms of cognitive impairment in MSA might be different with DLB and PDD. Given that the various involvements of cortico-subcortical pathways in MSA,7 aberrant circuits projecting from striato-nigral system to frontal lobe may be related. Although frontal lobe dysfunction was prominent in this patient, metabolism in both parietal lobe and posterior cingulate gyrus was lower than that of frontal lobe. Given that the [18F]-FDG PET findings are similar to the pattern of AD,8 the pathology of AD such as amyloid plaque or tau-positive neurofibrillary tangle may contribute to cognitive impairment related with MSA. Decreased level of beta amyloid peptide 42 in CSF and tau protein observed from autopsy in MSA patients may support this hypothesis.9,10
- We report a MSA patient who has developed cerebellar ataxia after diagnosed with dementia. This case may suggest that the clinical diagnosis of MSA could not be excluded by the presence of dementia.
Discussion
Figure 1.Serial brain MRI findings of the patient. A: The initial T1-weighted sagittal image showed atrophy of cerebellum. B and C: And T2-weighted axial image showed no definite abnormality. D, E and F: After 2 years, brain MRI showed progression of atrophy of cerebellum, pons, midbrain and middle cerebellar peduncle. MRI: magnetic resonance imaging.
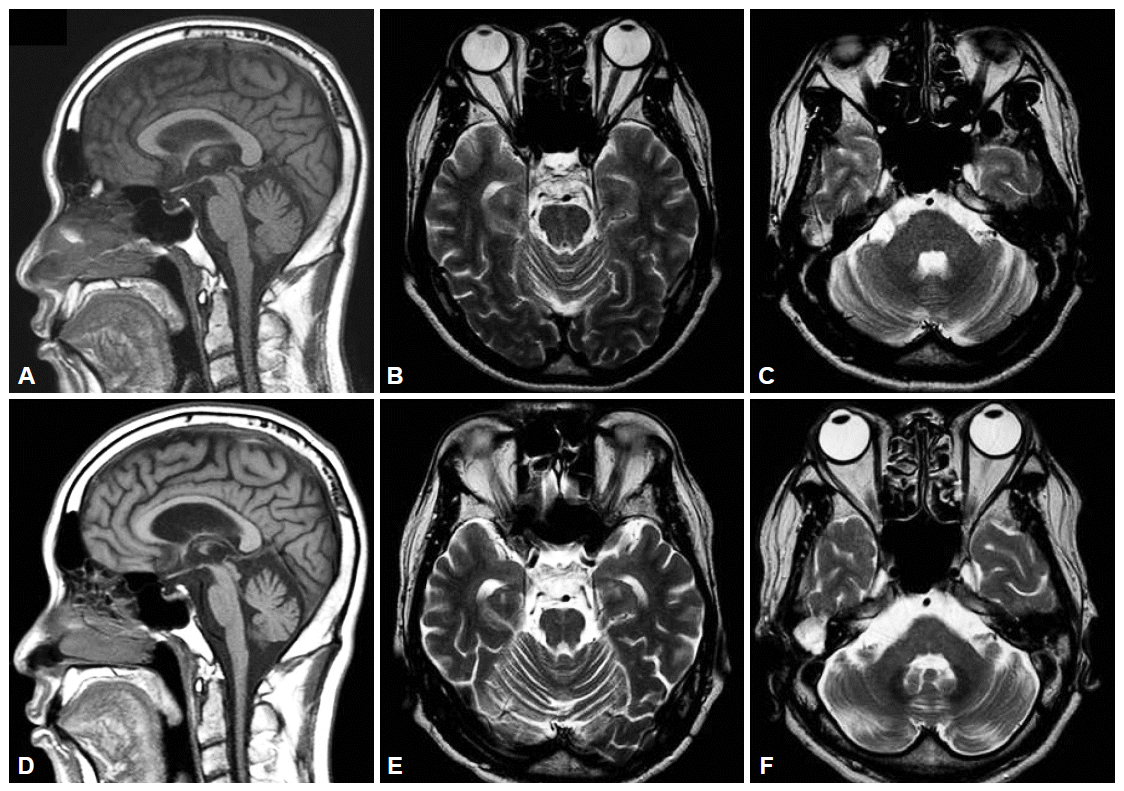
Figure 2.Brain [18F]-FDG PET images of the patient. Brain [18F]-FDG PET showed mildly decreased FDG uptake in the both posterior parietal lobe (A, white arrow). And also there were markedly decreased FDG uptake in both bagal ganglia and cerebellum (B and C). The sagittal image revealed mildly deceased FDG uptake in posterior cingulated area (D, empty arrow). PET: positron emission tomography, FDG: 18F-fluorodeoxyglucose.
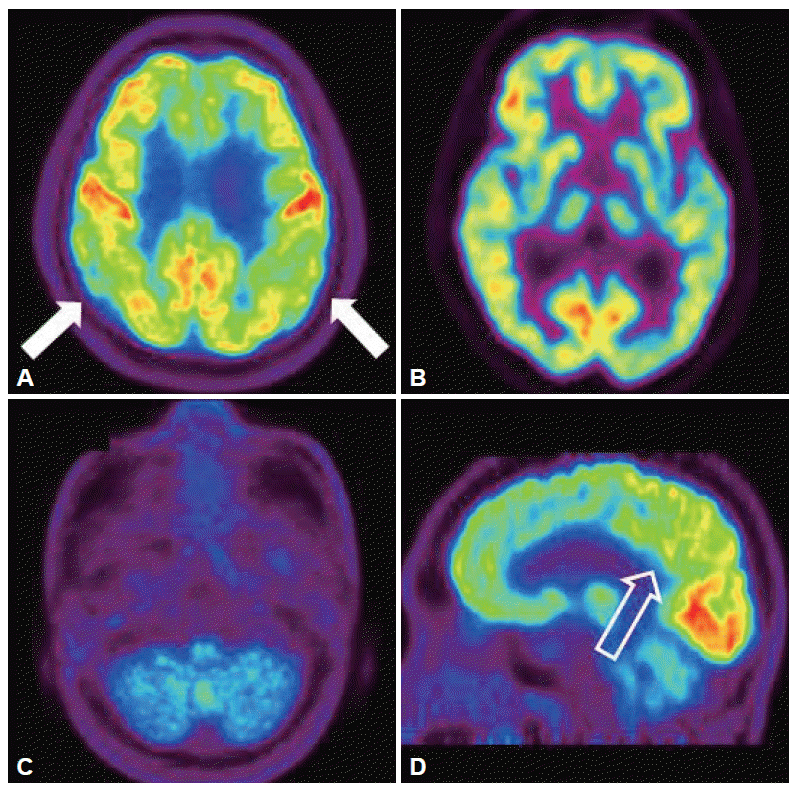
Table 1.Result of neuropsychological test
- 1. Balas M, Balash Y, Giladi N, Gurevich T. Cognition in multiple system atrophy: neuropsychological profile and interaction with mood. J Neural Transm 2010;117:369–375.ArticlePubMed
- 2. Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670–676.ArticlePubMedPMC
- 3. Kitayama M, Wada-Isoe K, Irizawa Y, Nakashima K. Assessment of dementia in patients with multiple system atrophy. Eur J Neurol 2009;16:589–594.ArticlePubMed
- 4. Chang CC, Chang YY, Chang WN, Lee YC, Wang YL, Lui CC, et al. Cognitive deficits in multiple system atrophy correlate with frontal atrophy and disease duration. Eur J Neurol 2009;16:1144–1150.ArticlePubMed
- 5. Kawai Y, Suenaga M, Takeda A, Ito M, Watanabe H, Tanaka F, et al. Cognitive impairments in multiple system atrophy: MSA-C vs MSA-P. Neurology 2008;70(16 Pt 2):1390–1396.ArticlePubMed
- 6. Kao AW, Racine CA, Quitania LC, Kramer JH, Christine CW, Miller BL. Cognitive and neuropsychiatric profile of the synucleinopathies: Parkinson disease, dementia with Lewy bodies, and multiple system atrophy. Alzheimer Dis Assoc Disord 2009;23:365–370.ArticlePubMedPMC
- 7. Watanabe H, Saito Y, Terao S, Ando T, Kachi T, Mukai E, et al. Progression and prognosis in multiple system atrophy: an analysis of 230 Japanese patients. Brain 2002;125(Pt 5):1070–1083.ArticlePubMed
- 8. Bohnen NI, Djang DS, Herholz K, Anzai Y, Minoshima S. Effectiveness and safety of 18F-FDG PET in the evaluation of dementia: a review of the recent literature. J Nucl Med 2012;53:59–71.ArticlePubMed
- 9. Holmberg B, Johnels B, Blennow K, Rosengren L. Cerebrospinal fluid Abeta42 is reduced in multiple system atrophy but normal in Parkinson’s disease and progressive supranuclear palsy. Mov Disord 2003;18:186–190.ArticlePubMed
- 10. Piao YS, Hayashi S, Hasegawa M, Wakabayashi K, Yamada M, Yoshimoto M, et al. Co-localization of alpha-synuclein and phosphorylated tau in neuronal and glial cytoplasmic inclusions in a patient with multiple system atrophy of long duration. Acta Neuropathol 2001;101:285–293.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
Citations to this article as recorded by 
- Mitochondrial dysfunction and altered ribostasis in hippocampal neurons with cytoplasmic inclusions of multiple system atrophy
Norihisa Maeda, Hiroyuki Honda, Satoshi O. Suzuki, Naoki Fujii, Jun‐ichi Kira, Toru Iwaki
Neuropathology.2018; 38(4): 361. CrossRef - Mechanisms and prevention of sudden death in multiple system atrophy
Takayoshi Shimohata, Naotaka Aizawa, Hideaki Nakayama, Hiroshige Taniguchi, Yasuyoshi Ohshima, Hitoshi Okumura, Tetsuya Takahashi, Akio Yokoseki, Makoto Inoue, Masatoyo Nishizawa
Parkinsonism & Related Disorders.2016;[Epub] CrossRef - Fragile X-associated tremor/ataxia syndrome: phenotypic comparisons with other movement disorders
Erin E. Robertson, Deborah A. Hall, Andrew R. McAsey, Joan A. O’Keefe
The Clinical Neuropsychologist.2016; 30(6): 849. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite