 E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 10(2); 2017 > Article
-
Case Report
Progressive Supranuclear Gaze Palsy with Predominant Cerebellar Ataxia: A Case Series with Videos - Zheyu Xu1, Tchoyoson C.C. Lim2, Wing Lok Au1, Louis C.S. Tan1
-
Journal of Movement Disorders 2017;10(2):87-91.
DOI: https://doi.org/10.14802/jmd.16059
Published online: April 18, 2017
1Department of Neurology, National Neuroscience Institute, Singapore, Singapore
2Department of Neuroradiology, National Neuroscience Institute, Singapore, Singapore
- Corresponding author: Tan Chew Seng Louis, FRCP, Department of Neurology, National Neuroscience Institute, 11 Jalan Tan Tock Seng, Singapore 308433, Singapore / Tel: +65-6357-7153 / Fax: +65-6256-4755 / E-mail: louis.tan.c.s@singhealth.com.sg
Copyright © 2017 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Progressive supranuclear palsy (PSP) with predominant cerebellar ataxia (PSP-C) is a rare phenotype of PSP. The clinical and radiological features of this disorder remain poorly characterized. Through a retrospective case series, we aim to characterize the clinical and radiological features of PSP-C. Four patients with PSP-C were identified: patients who presented with prominent cerebellar dysfunction that disappeared with the progression of the disease. Supranuclear gaze palsy occurred at a mean of 2.0 ± 2.3 years after the onset of ataxia. Mild cerebellar volume loss and midbrain atrophy were detected on brain imaging, which are supportive of a diagnosis of PSP. Videos are presented illustrating the co-existence of cerebellar signs and supranuclear gaze palsy and the disappearance of cerebellar signs with disease progression. Better recognition and the development of validated diagnostic criteria would aid in the antemortem recognition of this rare condition.
- Four patients (3 males and 1 female) with PSP-C were identified. The average age of onset was 58.3 ± 3.3 years with a mean follow-up of 5 years. None of the patients had a family history of neurodegenerative disease. The diagnostic workup included spinocerebellar ataxia (SCA) types 1, 2, 3, and 6, dentatorubral-pallidoluysian atrophy, CTs of the chest, abdomen and pelvis, vitamin B12 and vitamin E levels, ceruloplasmin, lactate levels, syphilis, and HIV tests, and anti-glutamic acid decarboxylase and anti-endomysial antibodies, which were negative. The average time from onset to falls, supranuclear gaze palsy and cognitive impairment was 2.0 ± 2.3, 4.3 ± 1.7, and 3.5 ± 2.4 years, respectively. None of the patients developed dysautonomia, and 2 patients underwent further urological evaluation, which did not detect a neurogenic bladder. None of the patients had alien limb syndrome or cortical sensory deficits on examination.
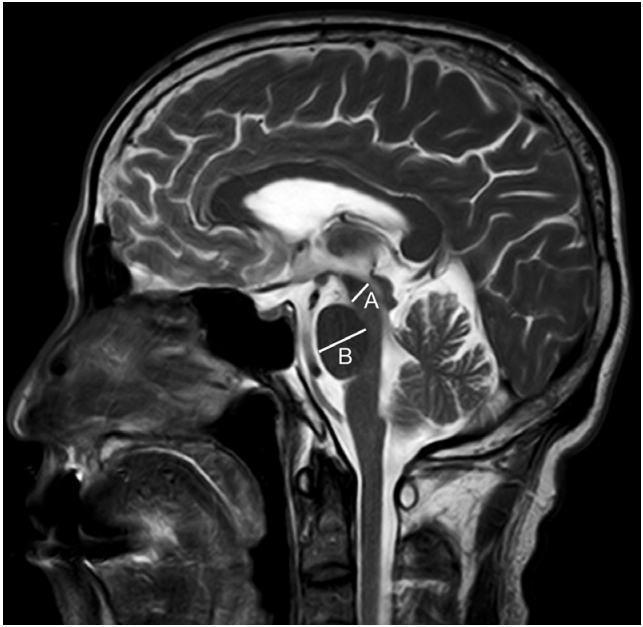
- Sagittal sequences on brain MRI were available for 2 patients (cases 1 and 4), which showed midbrain atrophy: midbrain measurements were 8.2 mm in both patients, with midbrain-to-pontine ratios of 0.44 and 0.48 (Figure 1). In the 2 remaining patients where sagittal MRI brain sequences were unavailable, mild cerebellar volume loss was detected. The hot cross bun sign, heterogeneous regions in the cerebellar dentate nucleus, increased signal within the superior cerebellar peduncle, focal frontal or temporoparietal atrophy and other vascular lesions were absent in all patients. The patients are summarized in Table 1.
- Case 1
- This 60-year-old Chinese male presented with gait difficulties and frequent falls at the age of 56. Examination findings included dysarthria of speech, bilateral limb ataxia and dysdiadochokinesia with postural instability. At age 59, the patient developed a preference for sweet foods. At age 60, supranuclear gaze palsy was detected with marked cognitive impairment and apathy [Mini-Mental State Examination (MMSE) 22/30, Montreal Cognitive Assessment (MOCA) 14/30]. Examination findings revealed frontalis hyperactivity and square wave jerks with marked rigidity that masked the initial cerebellar signs. The patient scored 56 on the Unified Parkinson’s Disease Rating Scale (UPDRS) Part III. The diagnosis was revised to PSP-C.
- Case 2
- This 67-year-old Chinese female first presented with a 6-month history of frequent falls at age 63. Findings included dysarthria of speech, limb ataxia and a wide-based gait. At age 64, cognitive impairment (MOCA 18/30) and pseudobulbar affect was evident. The patient was given a diagnosis of multiple system atrophy-cerebellar (MSA-C) in view of possible urge incontinence. Two years later, supranuclear gaze palsy was detected. Subsequent examination showed frontalis hyperactivity, restriction of the vertical upgaze, marked nuchal rigidity, and minimal limb ataxia associated with frontal disinhibition and inappropriate laughter. She scored a 69 on Part III of the UPDRS. The diagnosis was revised to PSP-C (Supplementary Video 1 in the Online-only Data Supplement).
- Case 3
- This 62-year-old Chinese male presented with a 3-year history of right upper limb incoordination progressing to gait difficulty and falls at age 59. Cognitive impairment was apparent at onset. The MMSE score was 24/30. Examination findings included masked facies, scanning speech, gaze-evoked nystagmus, limb ataxia, dysdiadochokinesia with a wide-based gait and poor postural reflexes. The patient was bradykinetic with freezing when walking and was diagnosed with MSA-C. A trial of levodopa did not result in symptomatic benefit and was stopped. At age 60, supranuclear gaze palsy was detected. The last recorded UPDRS Part III was 49. The patient died at age 62.
- Case 4
- This 64-year-old Chinese male presented with right upper limb incoordination at age 58. At age 62, he had slowed horizontal saccades, bilateral limb ataxia, a broad-based ataxic gait and mild right-sided bradykinesia without rigidity. At age 63, a change in personality with the tendency to anger and cognitive impairment was noted. At age 64, examination findings showed restricted eye movements with supranuclear gaze palsy, bilateral dysmetria, grade 1 rigidity and grade 2 bradykinesia (Supplementary Video 2 in the Online-only Data Supplement). The patient scored a 36 on the UPDRS Part III, 26/30 on the MMSE and 14/30 on the MOCA. The midbrain was atrophied (measuring 8.2 mm) (Table 1, Figure 1), and the midbrain-to-pons ratio was 0.48. The diagnosis was revised to PSP-C.
CASE REPORT
- We present a case series of 4 patients with PSP-C, with videos demonstrating the co-existence of ataxia and the typical clinical findings of PSP.
- In our series, all patients presented with ataxia, followed by the later development of supranuclear gaze palsy without dysautonomia and an absence of the hot cross bun sign on imaging, which closely corresponds to the proposed diagnostic criteria of PSP-C [6]. However, early falls did not occur in 2 of our patients, although the tendency to fall had occurred early. Limitations to the current NINDS-SPSP criteria for the diagnosis of PSP have been recognized, and there has been a move to redefine postural instability instead as the tendency to fall. Further refinement of the proposed diagnostic criteria of PSPC is likely considering a recently described case in which dysautonomia was present [2].
- We were able to demonstrate a reduced midbrain measurement of < 9.35 mm and a midbrain-to-pons ratio of less than 0.52 in 2 patients, which has been shown to be 100% specific for the diagnosis of PSP in autopsy-proven cases [7]. Mild cerebellar volume loss was present in the remaining 2 cases at 0.5 and 3 years after the onset of disease. Cerebellar atrophy had previously only been described in the advanced stage of disease [5], and our limited number of cases may not be generalizable.
- The average age of onset in our patients was 58.3 ± 3.3 years, which is a decade younger than previously reported [8]. Age of onset has been thought to be a distinguishing feature of PSP-C compared to MSAC [8]. Our findings suggest that PSP-C and MSA-C could have similar ages of onset and that age is not a useful differentiating factor.
- The proposed diagnostic criteria have suggested that PSP-C has a slowly progressive course [6]. However, 1 patient exhibited rapid disease progression over 6 years, with a short time to death. Furthermore, the time from onset to the occurrence of falls, cognitive impairment and supranuclear gaze palsy in our patients was rapid and closely follows the trajectory of the Richardson-Steele phenotype of PSP, which has the worst prognosis [9]. The mean disease duration of PSP-C from previously reported cases have ranged from 3 to 11 years with a mean of 6 years [2-5], which suggests that PSP-C does not have a benign course.
- In our series, the average time of onset of supranuclear palsy was 4.3 ± 1.7 years, with one patient who developed supranuclear gaze palsy 6 years after the onset of ataxia. Supranuclear gaze palsy was detected in 8 of the 9 autopsy-proven Japanese cases, with 3 that occurred within 2 years of onset [5]. In contrast, supranuclear gaze palsy was detected antemortem in only 1 of the 5 Western cases despite a long duration of disease [4]. These patients had ataxia as the only clinical finding. An unknown proportion of patients with adult-onset cerebellar ataxia could consequently be cases of PSP-C that do not develop supranuclear palsy antemortem. Proposed diagnostic criteria should reflect the wide variability in the onset of supranuclear gaze palsy, rather than restricting the case definition to patients with supranuclear gaze palsy occurring within 2 years of onset [6].
- With disease progression, limb ataxia was difficult to detect clinically. This disappearance of cerebellar signs has been previously described [10] and could be due to the subsequent development of marked rigidity and bradykinesia that leads to masking of the co-existing ataxia due to a slower performance of motor tasks.
- We were able to characterize the PSP-C phenotype at the early stages; however, the limitation of our study is the absence of neuropathological confirmation. Other differentials to consider include MSA-C and spinocerebellar ataxias. Further longitudinal studies with autopsy confirmation will be required.
- In conclusion, we present 4 patients in early- and mid-stage PSP-C disease. Clinicians should be vigilant to this diagnostic possibility as supranuclear gaze palsy may manifest years after the onset of ataxia. However, further neuropathological confirmation of such patients will be necessary to define this rare phenotype to allow better antemortem detection.
DISCUSSION
Supplementary Materials
Supplementary Video Legends
Supplementary Video Legends
- The research is supported by the Singapore National Research Foundation under its Translational and Clinical Research Flagship Programme (TCR12dec010) and administered by the Singapore Ministry of Health’s National Medical Research Council.
Acknowledgments
| Sex | Age of onset (years) | Symptoms at onset | Time to onset of falls/years | Time from onset to detection of supranuclear gaze palsy/years | Time from onset to development of cognitive impairment/years | MMSE | MOCA | Initial diagnosis | Time from onset to brain imaging/years |
Brain imaging findings |
Time from onset to walking aid use/months | Last MRS score | Time from onset to death/years | Length of follow up/years | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Midbrain/pons measurement/mm | Midbrain to pons ratio | ||||||||||||||||
| 1 | M | 56 | Unsteady gait, falls | 0 | 4 | 4 | 22 | 14 | Cerebellar degeneration | 0.5 | No cerebellar volume loss | 8.2/18.6 | 0.44 | 36 | 4 | NA | 4 |
| 2* | F | 63 | Unsteady gait, falls | 0 | 2 | 0 | NA | 18 | MSA-C | 0.5 | Mild cerebellar volume loss | NA† | NA† | 4 | 5 | NA | 4 |
| 3 | M | 56 | Upper limb clumsiness | 4 | 5 | 5 | 24 | NA | MSA-C | 3.0 | Mild cerebellar volume loss | NA† | NA† | 48 | 6 | 6 | 6 |
| 4* | M | 58 | Unsteady gait, clumsiness | 4 | 6 | 5 | 26 | 14 | Cerebellar degeneration | 1.0 | No cerebellar volume loss | 8.2/17.2 | 0.48 | 42 | 4 | NA | 6 |
| Total (mean ± SD) | 58.3 ± 3.3 | 2.0 ± 2.3 | 4.3 ± 1.7 | 3.5 ± 2.4 | 24 ± 2.0 | 15.3 ± 2.3 | 32.5 ±19.6 | 5‡ | |||||||||
* cases 2 and 4 have accompanying videos,
† midbrain and pontine measurements could not be obtained as the sagittal sequences on brain MRI were unavailable,
‡ median.
NA: not applicable, UPDRS: Unified Parkinson’s Disease Rating Scale, MMSE: Mini-Mental State Examination, MOCA: Montreal Cognitive Assessment, MRS: Modified Rankin Score, MSA-C: multiple system atrophy-cerebellar, SD: standard deviation, PSP-C: progressive supranuclear palsy with pedominant cerebellar ataxia.
- 1. Kanazawa M, Shimohata T, Toyoshima Y, Tada M, Kakita A, Morita T, et al. Cerebellar involvement in progressive supranuclear palsy: a clinicopathological study. Mov Disord 2009;24:1312–1318.ArticlePubMed
- 2. Mensikova K, Tuckova L, Ehrmann J, Kanovsky P. Unusual phenotype of pathologically confirmed progressive supranuclear palsy with autonomic dysfunction and cerebellar ataxia: case report. Medicine (Baltimore) 2016;95:e5237.ArticlePubMedPMC
- 3. Lee MJ, Lee JH, Kim BK, Lee JH, Lee YM, Kim SJ, et al. An autopsy confirmed case of progressive supranuclear palsy with predominant cerebellar ataxia. J Neurol 2016;263:2540–2543.ArticlePubMedPDF
- 4. Koga S, Josephs KA, Ogaki K, Labbé C, Uitti RJ, Graff-Radford N, et al. Cerebellar ataxia in progressive supranuclear palsy: an autopsy study of PSP-C. Mov Disord 2016;31:653–662.ArticlePubMedPMC
- 5. Shimohata T, Kanazawa M, Yoshida M, Saito Y, Iwai K, Yasuda T, et al. Clinical and imaging findings of progressive supranuclear palsy with predominant cerebellar ataxia. Mov Disord 2016;31:760–762.ArticlePubMed
- 6. Shimohata T, Kanazawa M, Takahashi H, Nishizawa M. Clinicopathological features and diagnostic criteria for progressive supranuclear palsy with predominant cerebellar ataxia [abstract]. Mov Disord 2015;30 Suppl 1:847.
- 7. Massey LA, Jäger HR, Paviour DC, O’Sullivan SS, Ling H, Williams DR, et al. The midbrain to pons ratio: a simple and specific MRI sign of progressive supranuclear palsy. Neurology 2013;80:1856–1861.ArticlePubMedPMC
- 8. Kanazawa M, Tada M, Onodera O, Takahashi H, Nishizawa M, Shimohata T. Early clinical features of patients with progressive supranuclear palsy with predominant cerebellar ataxia. Parkinsonism Relat Disord 2013;19:1149–1151.ArticlePubMed
- 9. Respondek G, Stamelou M, Kurz C, Ferguson LW, Rajput A, Chiu WZ, et al. The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Mov Disord 2014;29:1758–1766.ArticlePubMed
- 10. Iwasaki Y, Mori K, Ito M, Tatsumi S, Mimuro M, Yoshida M. An autopsied case of progressive supranuclear palsy presenting with cerebellar ataxia and severe cerebellar involvement. Neuropathology 2013;33:561–567.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
- Central nystagmus in progressive supranuclear palsy: A neglected clinical feature?
Maja Klarendic, Manja Hribar, Nina Bozanic Urbancic, Nina Zupancic, Milica G. Kramberger, Maja Trost, Saba Battelino, Diego Kaski, Maja Kojovic
Parkinsonism & Related Disorders.2021; 84: 15. CrossRef - “Parkinson’s disease” on the way to progressive supranuclear palsy: a review on PSP-parkinsonism
Ján Necpál, Miroslav Borsek, Bibiána Jeleňová
Neurological Sciences.2021; 42(12): 4927. CrossRef - Progressive Supranuclear Palsy with Predominant Cerebellar Ataxia
Shoichiro Ando, Masato Kanazawa, Osamu Onodera
Journal of Movement Disorders.2020; 13(1): 20. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite
