 E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 10(3); 2017 > Article
-
Case Report
Oculodentodigital Dysplasia Presenting as Spastic Paraparesis: The First Genetically Confirmed Korean Case and a Literature Review - Kye Won Park1, Ho-Sung Ryu1, Juyeon Kim2, Sun Ju Chung1
-
Journal of Movement Disorders 2017;10(3):149-153.
DOI: https://doi.org/10.14802/jmd.17050
Published online: September 22, 2017
1Department of Neurology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
2Department of Neurology, Metro Hospital, Anyang, Korea
- Corresponding author: Sun Ju Chung, MD, PhD, Department of Neurology, Asan Medical Center, University of Ulsan College of Medicine, 88 Olympic-ro 43-gil, Songpa-gu, Seoul 05505, Korea / Tel: +82-2-3010-3440 / Fax: +82-2-474-4691 / E-mail: sjchung@amc.seoul.kr
Copyright © 2017 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Oculodentodigital dysplasia (ODDD) is a rare autosomal dominant inherited disease caused by mutations of the human gap junction alpha 1 gene, which encodes the protein Connexin-43. Patients with ODDD may present with neurological deficits with a typical pleiotropic combination of characteristic craniofacial, ophthalmological, phalangeal, and dental anomalies. In this report, we describe the first genetically confirmed Korean ODDD patient, who presented with spastic paraparesis. We will also review the neurological aspects of ODDD as reported in the literature.
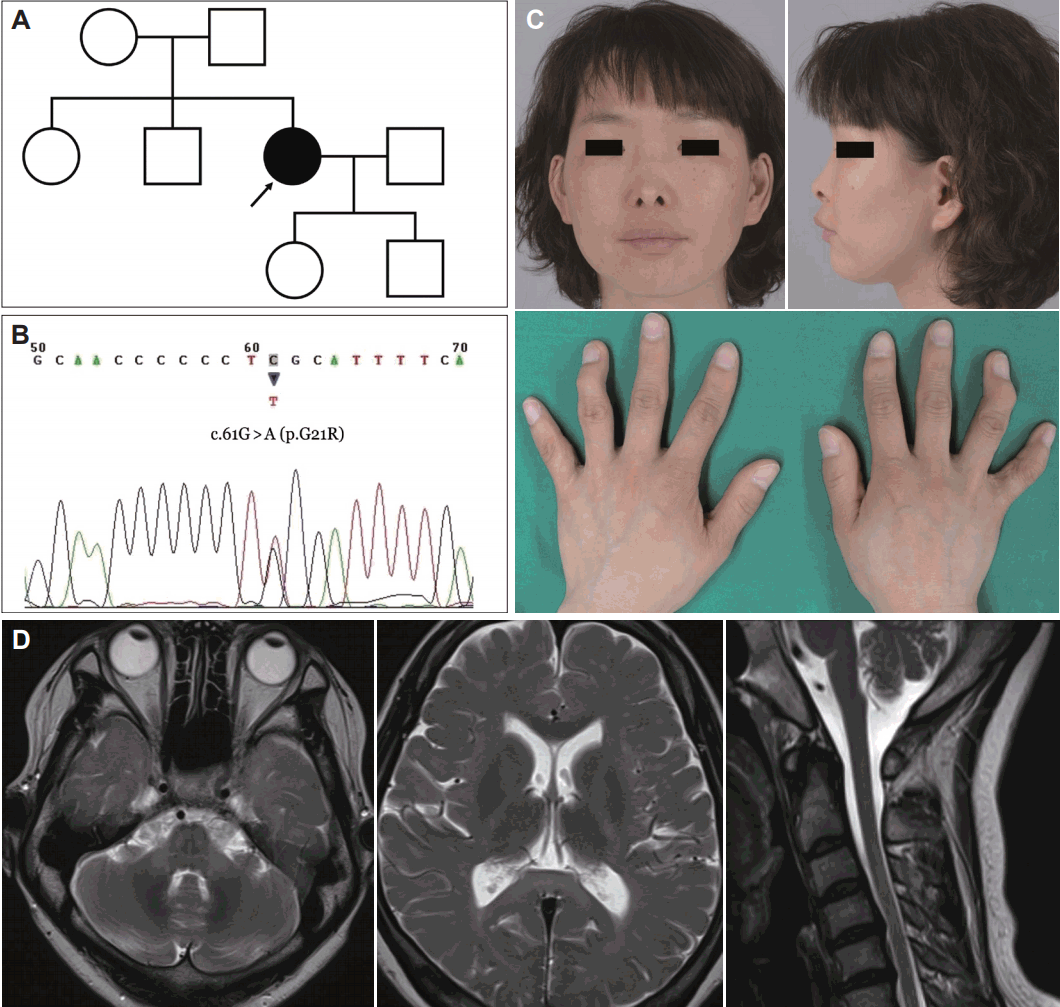
- A 40-year-old female visited our neurology clinic because of gait disturbance. She was born to term with syndactyly of her bilateral fourth and fifth finger (syndactyly type III), which was surgically corrected. There was no developmental delay, but she was always in last place in her class in track and field. She had been working as a salesperson since graduating from high school. She began to feel stiffness in her legs in her mid-30s. She also suffered from urinary frequency and incomplete voiding of urine. These symptoms worsened over the following five years. Her father had suffered intracerebral hemorrhage 2 years prior but did not manifest with any gait problems. She was married and had two children, but none of her siblings, children, or nephews had a gait problem (Figure 1A). There was no history of seizures.
- Upon physical examination, she was found to have microphthalmia with small sunken eyes, thin nose, hypoplastic ale nasi, and bilateral epicanthic folds. In her hands, there were scars from previous surgeries for syndactyly type III and campylodactyly (fixed flexion of deformity of the fingers and toes) of bilateral fourth fingers (Figure 1C). Her skin and hair were brittle and dry.
- Upon neurological examination, there was spasticity, generalized hyperreflexia, Babinski’s sign, and ankle clonus on her lower extremities. She also showed bilateral Hoffman’s sign. Scissor gait was observed. Her mini-mental status examination score was 30 out of 30. On brain MRI, there were diffuse high signal intensities of bilateral cerebral white matter from the juxta-cortical area to the deep white matter, extending to the brainstem. There was diffuse atrophy of the brain, brainstem and spinal cord (Figure 1D). She also complained of urinary frequency and nocturia. She had 200–300 cc of residual urine after urination and needed intermittent catheterization. Her visual and auditory functions were unremarkable. Nerve conduction studies of her bilateral median, ulnar, peroneal, and tibial nerves were also unremarkable. Transcranial magnetic motor-evoked potentials from her bilateral tibialis anterior muscles were normal after lumbar stimulation but were unobtainable after cortical stimulation, suggesting central conduction block in the corticospinal tract.
- Sanger sequencing of the patient’s GJA1 gene using her peripheral blood samples identified the heterozygous missense mutation c.61G > A (Figure 1B), which had been previously reported as a pathogenic mutation [1,4]. No mutations were found in the SPG3A or SPG4 genes. Clonazepam, baclofen, and trihexyphenidyl were administered for symptomatic relief, which resulted in mild improvement of spasticity of her lower extremities.
CASE REPORT
- This patient presented in our movement clinic with spastic gait disturbance. Although ODDD is not the first-line differential diagnosis for adult onset spastic paresis due to its extreme rarity, her distinct craniofacial and digital features led us to perform the diagnostic gene test. The case showed the typical features of ODDD, including craniofacial dysmorphisms, syndactyly, spastic gait disturbance, and abnormal high signal intensities in the bilateral cerebral white matter on brain MRI. To the best of our knowledge, she is the first genetically confirmed ODDD case in Korea. In previous reports, affected individuals have been mostly Caucasians [4]. Since the causative gene was identified in 2003, cases from East Asia have been accumulating, including our patient [5-7].
- ODDD is caused by the mutation in the GJA1 gene that encodes the protein Cx43, which is a member of the gap junction family protein. In the central nervous system, Cx43 gap junction channels are mostly found in astrocytes [2]. Defective Cx43 proteins by the mutation in GJA1 gene results in non-functional gap junctions and aberrant signaling in astrocytes [2,8]. At present, according to the HGMD® Public database, 2017. 6, there are 89 different mutations that are associated with ODDD [9]. Among the reported mutations, more than 30 have been associated with neurological involvement. The mutations, neurological symptoms, origins, and obtained literature are summarized in Table 1.
- The prevalence of ODDD has been reported to be less than 1/1,000,000 [2]. This syndrome is highly penetrant with enormous intra- and interfamilial phenotypic variability [4]. The constellation of symptoms of ODDD is composed of typical craniofacial dysmorphisms, neurological manifestations, ophthalmological anomalies, dental anomalies, malformation of extremities, brittle hair/skin, and other miscellaneous features [3,4]. The craniofacial dysmorphisms (thin nose, hypoplastic alae nasi, small nares, and cleft lip and palate), malformation of the extremities (syndactyly, campylodactyly, and clinodactyly), dental features (microdontia and enamel hypoplasia), and ophthalmological anomalies (microphthalmia, microcornia, iris abnormalities, cataract, and glaucoma) are the most common, being observed in 92, 80, 70, and 68% of cases, respectively [1-4].
- Neurological manifestations of ODDD include upper motor neuron signs including spasticity, neurogenic bladder, epilepsy, mental retardation, and abnormal brain imaging findings [3]. According to Paznekas et al. [4], neurological manifestations are seen in 30% of affected families. This number may have been underestimated for several reasons. First, neurological ODDD symptoms usually manifest later in life, typically in the third to fourth decades, whereas the craniofacial and digital findings are already evident at the time of birth. Second, neurological symptoms can be easily neglected if the diagnosis of ODDD is made by physicians with other specialties, especially when the symptoms are subtle. Moreover, there are cases that have not yet manifested neurological symptoms but have abnormal brain imaging findings [7,10]. Their neurological symptoms may become evident later in life. Thus, a thorough, multi-disciplinary, longitudinal approach for each patient is warranted.
- The phenotypes of neurological manifestation may vary, as seen in Table 1. Meanwhile, in those who underwent brain imaging, they exhibited similar abnormal imaging findings, such as symmetrical generalized white matter signal changes extending to the brainstem, atrophy of the brainstem and cerebellum, and calcification of the basal ganglia and dentate nucleus [4,7,10]. This finding may reflect the degeneration of the corticospinal tract and related regions caused by the aberrant signaling of gap junctions of astrocytes.
- In this report, we describe a new Korean ODDD case who presented spastic paraparesis. ODDD is rare and associated with variable multi-systemic manifestations. Thus, ODDD poses a considerable diagnostic challenge. Appropriate brain imaging, as well as detailed neurological examination, is necessary for each patient to evaluate the extent of neurological involvement.
DISCUSSION
Supplementary Materials
- This work was supported by a grant of the Korea Healthcare Technology R & D Project, Ministry of Health and Welfare, Republic of Korea (HI14C2206).
Acknowledgments
| Protein change | Nucleotide change | Neurological symptoms | Origin | Age at diagnosis | Reference (years) |
|---|---|---|---|---|---|
| p.D3N | n.d. | UMN, NB, Uthoff's sign | n.d. | 14 | Churko et al. (2011) |
| p.L11F | c.31C > T | hyperactivity disorder, sensorineural hearing loss | Poland | 3.5 | Jamsheer et al. (2009) |
| p.Y17S | c.50A > C | UMN, NB | Canada | 22‒61 | Paznekas et al. (2009) |
| pS18P | c.52T > C | bilateral optic atrophy, MRI WMC | France | 5 | Alao et al. (2009) |
| p.G21R | c.61G > A | UMN | n.d. | 6 | Paznekas et al. (2009) |
| p.G22E | c.65G > A | UMN, MRI WMC, headache, Tremor | USA | 14 | Paznekas et al. (2009) |
| p.K23T | c.68A > C | UMN, MRI WMC, tremor, delayed speech | USA | 12 | Paznekas et al. (2009) |
| p.W25C | c.77G > T | UMN, MRI WMC | Japan | 34 | Furata et al. (2012) |
| p.W25C | c.75G > T | MRI WMC without neurological deficit | India | 7 | Dwarakanathan et al. (2015) |
| p.R33X | c.97C > T | UMN, MRI WMC | Pakistan | 1‒3.5 | Paznekas et al. (2009) |
| p.A40V | c.119C > T | gait difficulty, NB | n.d. | 8‒48 | Paznekas et al. (2009) |
| p.Q42G | c.124G > C | UMN, bipolar disorder, calcification of basal ganglia and dentate nucleus | Italy | 59 | Tumminelli et al. (2016) |
| p.Q49P | c.146A > C | NB | Austria/Turkey | 9 | Paznekas et al. (2009) |
| p.R76S | c.226C > A | MRI WMC, Sz | USA | 0‒adult | Paznekas et al. (2009) |
| p.R76C | c.226C > T | Cerebral infarction due to cardiac anomaly | Japan | 0 | Izumi et al. (2013) |
| p.S86Y | c.257C > A | Mental retardation | Austria | 0 | Jamsheer et al. (2014) |
| p.L90V | c.268C > G | UMN, NB, Sz | Norway | 7‒62 | Paznekas et al. (2009) |
| p.H95R | c.284A > G | UMN, NB, MRI WMC | n.d. | Unknown‒47 | Paznekas et al. (2009) |
| p.V96A | c.287T > C | MRI WMC | Australia | 2 | Paznekas et al. (2009) |
| p.K102N | c.306G > C | NB | n.d. | 4‒27 | Paznekas et al. (2009) |
| p.L106P | c.317T > C | UMN, NB | n.d. | 60 | Paznekas et al. (2009) |
| p.L113P | c.338T > C | UMN, MRI WMC | n.d. | 35‒63 | Paznekas et al. (2009) |
| p.I130T | c.389T > C | UMN, NB, Sz, MRI WMC & atrophy of spinal cord | USA | 17‒71 | Amador et al. (2008) |
| p.K134E | c.400A > G | UMN, MRI WMC | Russia | 29 | Paznekas et al. (2009) |
| p.K134N | c.402G > T | UMN | n.d. | Unknown | Paznekas et al. (2009) |
| p.G138R | c.412G > C | UMN, NB, MRI WMC | England/Ireland | 11‒Adult | Paznekas et al. (2009) |
| p.G138S | c.412G > A | MRI WMC without neurological deficit | Japan | 22 | Kogame et al. (2014) |
| p.T154A | c.460A > G | UMN, NB, MRI WMC | n.d. | 10 | Paznekas et al. (2009) |
| p.T154N | c.461C > A | UMN, NB, tremor | n.d. | 15‒47 | Paznekas et al. (2009) |
| p.V216L | c.646G > T | UMN, NB, MRI WMC | Ireland | 2‒41 | Paznekas et al. (2009) |
| p.S220Y | c.659C > A | Developmental disorder | Germany | 3 | Wiest et al. (2006) |
| p.Y230CfsX6 | c.689_690delTA | Mental retardation | France | 22 | Alao et al. (2009) |
| p.G2fsX7, p.R101X* | c.6delT(p.G2fsX7), c.301C > T(p.R101X) | Mental retardation, Sz, massive calcification of basal ganglia and dentate nucleus, hypomyelination, atrophy | Poland | 15 | Jamsheer et al. (2010) |
| n.d. | n.d. | UMN, Sz | Iran | 11 | Barzegar et al. (2012) |
| n.d. | n.d. | UMN, immature behavior, tremor, MRI WMC | Iran | 22 | Shakiba et al. (2012) |
References are summarized in Supplementary Material.
* compound heterozygous.
GJA1: gap junction alpha 1, n.d: not documented, UMN: upper motor neuron signs including spasticity, hyperreflexia, pathological reflexes, NB: neurogenic bladder or bowel signs, MRI WMC: magnetic resonance imaging white matter changes, Sz: seizure.
- 1. Paznekas WA, Boyadjiev SA, Shapiro RE, Daniels O, Wollnik B, Keegan CE, et al. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am J Hum Genet 2003;72:408–418.ArticlePubMedPMC
- 2. De Bock M, Kerrebrouck M, Wang N, Leybaert L. Neurological manifestations of oculodentodigital dysplasia: a Cx43 channelopathy of the central nervous system? Front Pharmacol 2013;4:120.ArticlePubMedPMC
- 3. Loddenkemper T, Grote K, Evers S, Oelerich M, Stögbauer F. Neurological manifestations of the oculodentodigital dysplasia syndrome. J Neurol 2002;249:584–595.ArticlePubMed
- 4. Paznekas WA, Karczeski B, Vermeer S, Lowry RB, Delatycki M, Laurence F, et al. GJA1 mutations, variants, and connexin 43 dysfunction as it relates to the oculodentodigital dysplasia phenotype. Hum Mutat 2009;30:724–733.ArticlePubMed
- 5. Furuta N, Ikeda M, Hirayanagi K, Fujita Y, Amanuma M, Okamoto K. A novel GJA1 mutation in oculodentodigital dysplasia with progressive spastic paraplegia and sensory deficits. Intern Med 2012;51:93–98.ArticlePubMed
- 6. Izumi K, Lippa AM, Wilkens A, Feret HA, McDonald-McGinn DM, Zackai EH. Congenital heart defects in oculodentodigital dysplasia: report of two cases. Am J Med Genet A 2013;161A:3150–3154.ArticlePubMed
- 7. Kogame T, Dainichi T, Shimomura Y, Tanioka M, Kabashima K, Miyachi Y. Palmoplantar keratosis in oculodentodigital dysplasia with a GJA1 point mutation out of the C-terminal region of connexin 43. J Dermatol 2014;41:1095–1097.ArticlePubMed
- 8. Lai A, Le DN, Paznekas WA, Gifford WD, Jabs EW, Charles AC. Oculodentodigital dysplasia connexin43 mutations result in non-functional connexin hemichannels and gap junctions in C6 glioma cells. J Cell Sci 2006;119:532–541.ArticlePubMed
- 9. HGMD® Public. The Human Gene Mutation Databese. [cited 2017 June]. Available from: http://www.hgmd.cf.ac.uk/ac/index.php.
- 10. Dwarakanathan A, Bhat M, Gn S, Shetty S. Missense and deletion mutations in GJA1 causing oculodentodigital dysplasia in two Indian families. Clin Dysmorphol 2015;24:159–162.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
- Glial Connexins and Pannexins in the Healthy and Diseased Brain
Christian Giaume, Christian C. Naus, Juan C. Sáez, Luc Leybaert
Physiological Reviews.2021; 101(1): 93. CrossRef - Oculodentodigital Dysplasia: A Case Report and Major Review of the Eye and Ocular Adnexa Features of 295 Reported Cases
Virang Kumar, Natario L. Couser, Arti Pandya
Case Reports in Ophthalmological Medicine.2020; 2020: 1. CrossRef - Novel ocular findings in oculodentodigital dysplasia (ODDD): a case report and literature review
Zhirong Wang, Limei Sun, Panfeng Wang, Chonglin Chen, Aiyuan Zhang, Weiqing Wang, Xiaoyan Ding
Ophthalmic Genetics.2019; 40(1): 54. CrossRef - Oculodentodigital Dysplasia: A Hypomyelinating Leukodystrophy with a Characteristic MRI Pattern of Brain Stem Involvement
I. Harting, S. Karch, U. Moog, A. Seitz, P.J.W. Pouwels, N.I. Wolf
American Journal of Neuroradiology.2019; 40(5): 903. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite
