E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 11(3); 2018 > Article
-
Review Article
Clinical and Imaging Features of Multiple System Atrophy: Challenges for an Early and Clinically Definitive Diagnosis -
Hirohisa Watanabe1,2
 , Yuichi Riku2, Kazuhiro Hara2, Kazuya Kawabata2, Tomohiko Nakamura2, Mizuki Ito3, Masaaki Hirayama4, Mari Yoshida5, Masahisa Katsuno2, Gen Sobue1
, Yuichi Riku2, Kazuhiro Hara2, Kazuya Kawabata2, Tomohiko Nakamura2, Mizuki Ito3, Masaaki Hirayama4, Mari Yoshida5, Masahisa Katsuno2, Gen Sobue1 -
Journal of Movement Disorders 2018;11(3):107-120.
DOI: https://doi.org/10.14802/jmd.18020
Published online: August 9, 2018
1Brain and Mind Research Center, Nagoya University, Nagoya, Japan
2Department of Neurology, Nagoya University Graduate School of Medicine, Nagoya, Japan
3Toyota Kosei Hospital, Toyota, Japan
4Department of Pathophysiological Laboratory Science, Nagoya University Graduate School of Medicine, Nagoya, Japan
5Institute for Medical Science of Aging, Aichi Medical University, Nagakute, Japan
- Corresponding author: Hirohisa Watanabe, MD, PhD https://orcid.org/0000-0001-8553-8536 Department of Neurology, Nagoya University Graduate School of Medicine, 65 Tsurumai-cho, Syowa-ku, Nagoya 466-8550, Japan Tel: +81-52-744-2026 Fax: +81-52-731-8131 E-mail: nabe@med.nagoya-u.ac.jp
Copyright © 2018 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Multiple system atrophy (MSA) is an adult-onset, progressive neurodegenerative disorder. Patients with MSA show various phenotypes during the course of their illness, including parkinsonism, cerebellar ataxia, autonomic failure, and pyramidal signs. Patients with MSA sometimes present with isolated autonomic failure or motor symptoms/ signs. The median duration from onset to the concomitant appearance of motor and autonomic symptoms is approximately 2 years but can range up to 14 years. As the presence of both motor and autonomic symptoms is essential for the current diagnostic criteria, early diagnosis is difficult when patients present with isolated autonomic failure or motor symptoms/signs. In contrast, patients with MSA may show severe autonomic failure and die before the presentation of motor symptoms/signs, which are currently required for the diagnosis of MSA. Recent studies have also revealed that patients with MSA may show nonsupporting features of MSA such as dementia, hallucinations, and vertical gaze palsy. To establish early diagnostic criteria and clinically definitive categorization for the successful development of disease-modifying therapy or symptomatic interventions for MSA, research should focus on the isolated phase and atypical symptoms to develop specific clinical, imaging, and fluid biomarkers that satisfy the requirements for objectivity, for semi- or quantitative measurements, and for uncomplicated, worldwide availability. Several novel techniques, such as automated compartmentalization of the brain into multiple parcels for the quantification of gray and white matter volumes on an individual basis and the visualization of α-synuclein and other candidate serum and cerebrospinal fluid biomarkers, may be promising for the early and clinically definitive diagnosis of MSA.
- Multiple system atrophy (MSA) is defined as an adult-onset, rare, sporadic, and fetal neurodegenerative disease. The term MSA was proposed as encompassing three unrelated diseases: olivopontocerebellar atrophy (OPCA) [1], Shy-Drager syndrome (SDS) [2], and striatonigral degenerative disease (SND) [3] in 1969 by Graham and Oppenheimer [4]. In the same year, Takahashi et al. [5] independently reported similar concepts: 1) considerable common clinical and pathological features exist between OPCA and SDS, 2) both are nosologically allied conditions based on a detailed review of the literature, and 3) apparent striatonigral changes were similar to those implicated in the pathology of SND.
- Although there have been several discussions as to whether OPCA, SDS, and SND comprise a single disease or not, the discovery of the pathological hallmark, argyrophilic aggregates in the cytoplasm of oligodendrocytes, termed glial cytoplasmic inclusions (GCIs), resolved the dispute [6,7]. Subsequently, α-synuclein (α-syn) was proven to be the main component of GCIs in 1997 [8] and MSA was classified as an α-synucleinopathy together with Parkinson’s disease (PD) and dementia with Lewy bodies (DLB).
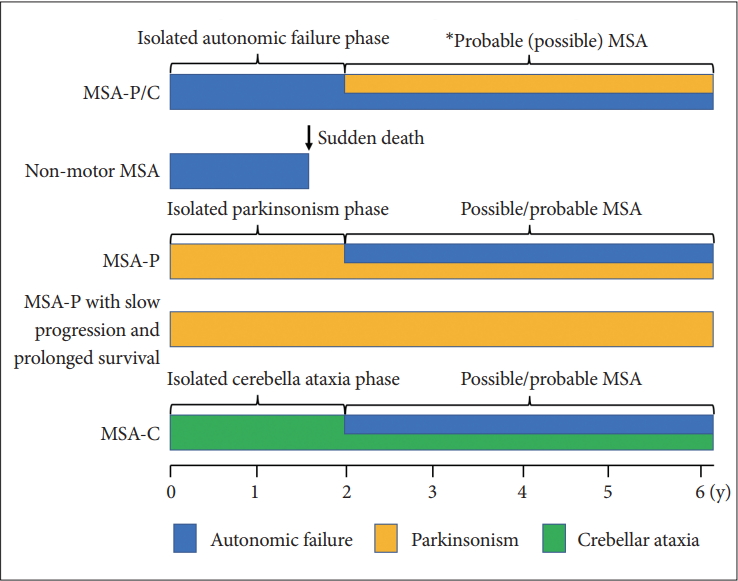
- Based on these findings, the first consensus statement on the diagnosis of MSA was proposed in 1998 [9]. In this statement, three levels of certainty were established: possible, probable, and definite. Definite MSA requires neuropathological confirmation. The combination of severe autonomic failure/urinary dysfunction plus poorly levodopa-responsive parkinsonism or cerebellar ataxia is required for a diagnosis of probable MSA. Patients with predominant parkinsonism are designated MSA-P and those with predominant cerebellar ataxia as MSA-C. It was also recommended to not use the term SDS.
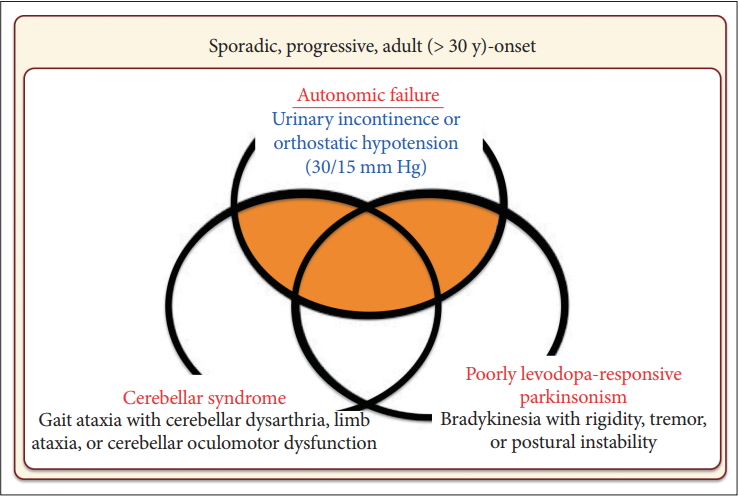
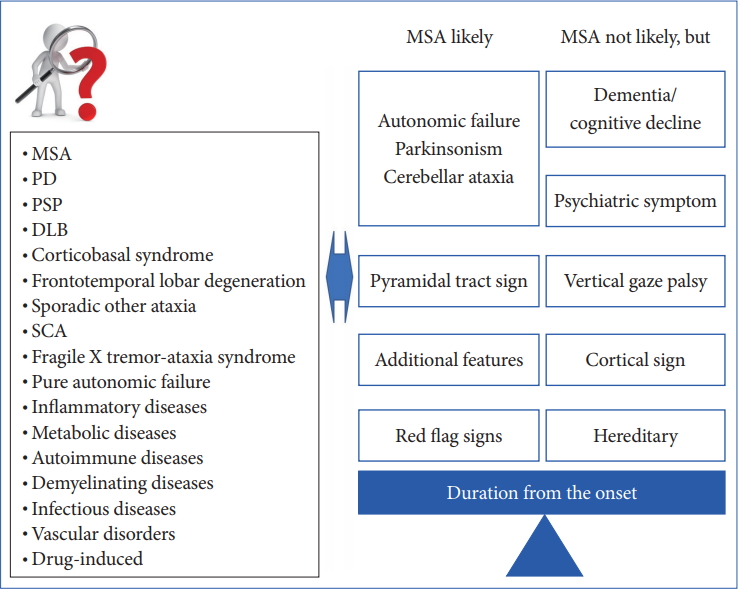
- In 2008, the second consensus statement was established [10]. These revised criteria also required rigorously defined autonomic failure and poorly levodoparesponsive parkinsonism or cerebellar ataxia for the diagnosis of probable MSA, similar to the first consensus statement (Figure 1). Possible MSA required parkinsonism or cerebellar ataxia and at least one feature suggesting autonomic dysfunction that did not fulfill the level required for probable MSA plus one other feature defined by clinical findings such as a Babinski sign with hyperreflexia, stridor, rapid progression rate, poor response to levodopa, and ataxia or characteristic neuroimaging abnormalities based on magnetic resonance imaging (MRI), 18fluoro-2-deoxyglucose positron emission tomography (FDG-PET), or presynaptic nigrostriatal dopaminergic PET/single photon emission computed tomography (SPECT) (Figure 2). Features supporting (red flags) and nonsupporting a diagnosis of MSA were also proposed (Table 1). Red flags are other clinical features based on the literature reviews and expert opinion that may raise the clinical suspicion of MSA, particularly for differentiating it from PD [11]. Classic pill-rolling rest tremor, clinically significant neuropathy, hallucinations not induced by medications, older age at onset (> 75 years), family history, dementia (as defined by the Diagnostic and Statistical Manual of Mental Disorders fourth edition, DSM-IV), and white matter lesions suggesting multiple sclerosis are nonsupporting features.
- Although several developments and refinements were incorporated in the second consensus criteria compared to the first, improvements in early diagnosis of MSA were limited (sensitivity of first clinical visit: 41% of possible MSA based on the second consensus criteria, 28% of possible MSA based on the first consensus criteria) [12]. Only 18% of patients fulfilled the second consensus criteria for probable MSA at the first clinical visit, although the positive predictive value was high and ranged from 86% to 100%. However, a more recent study showed that 62% of patients clinically diagnosed with MSA had received the correct diagnosis as shown at autopsy [13]. The presence of autonomic failure and cerebellar ataxia were the leading causes of misdiagnosis of DLB and progressive supranuclear palsy (PSP), respectively. Thus, a revision of the second consensus criteria for the early and clinically definitive diagnosis of MSA is urgently needed.
- In this article, we review the clinical features that can become obstacles for the early diagnosis of MSA and discuss the development of recent diagnostic biomarkers.
INTRODUCTION
- Time from onset to probable MSA
- The median time from initial symptom presentation to combined motor and autonomic dysfunction in MSA (probable MSA) is 2 years but ranges from 1 to 10 years [14]. Initial onset to the presence of concomitant motor and autonomic manifestations at 2, 4, and 6 years occurred in 57.4, 83.5, and 96.5% of patients, respectively. In other words, more than 40% of patients will visit the hospital with isolated motor or autonomic impairment and not be diagnosed as probable MSA within 2 years of onset. Since the median time from onset to death is 9 years but that from receiving a probable MSA diagnosis to death is 6 years, patients with probable MSA may be at severely advanced stages at diagnosis. Thus, it is important to focus on the clinical characteristics of the isolated autonomic failure or motor impairment (mono system atrophy) stage for early diagnosis.
- Isolated autonomic failure phase in MSA
- PD, DLB, and pure autonomic failure (PAF), classified as α-synucleinopathies, are the most important neurodegenerative diseases to consider in the differential diagnosis of MSA. Autonomic failure is commonly observed in all α-synucleinopathies. Severe autonomic failure that was classically considered an exclusion criterion for the diagnosis of PD [15] is one of the red flags but not an absolute exclusion criterion according to the Movement Disorders Society [16]. In the findings of a meta-analysis, orthostatic hypotension (OH) was observed in more than 30% of patients with PD [17]. Patients with DLB show autonomic failure more frequently than PD [18]. Interestingly, a prospective cohort study on PAF reported that 25 of 74 subjects with severe OH (34%) developed DLB (n = 13), PD (n = 6), or MSA (n = 6) within 4 years of follow up, supporting the idea that PD, DLB, PAF, and MSA can show isolated autonomic failure prior to reaching the full-blown stage [19].
- We should also pay attention to the nonmotor MSA phenotype [20]. Patients with nonmotor MSA are rare but present with severe autonomic failure leading to sudden death or respiratory failure in the absence of diagnostic motor symptoms/signs of MSA. In our autopsy series from 1976 to 2014, four of 161 patients fulfilled the criteria for nonmotor MSA [21]. The time from onset to death was 1.3 to 2 years. A pathological study showed severe involvement of the medullary serotonergic neurons, particularly in the nucleus raphe obscurus and nucleus of the ventrolateral medulla. A group of serotonergic neurons of the nucleus raphe obscurus projects to phrenic and other respiratory motoneurons and is intrinsically sensitive to levels of CO2 associated with respiratory dysfunction, such as impaired sensitivity to hypercapnia, suggesting an increased risk of sudden death [22-25]. Cell loss of serotonin-positive pre-Bötzinger complex cells as an essential part of the medullary respiratory network located in a circumscribed area of the ventrolateral medulla may also contribute to breathing disorders in MSA.
- Interestingly, patients with minimal change MSA with sudden death also showed marked involvement of medullary serotonergic neurons in our study [20]. Minimal change MSA presented with definite parkinsonism and dysautonomia but showed selective cell loss in the substantia nigra and locus coeruleus in combination with widespread GCI and astrogliosis [26]. Significant respiratory dysfunction and early OH were observed in minimal change MSA [21]. The clinicopathological findings of minimal change MSA were closely related to those of nonmotor MSA, suggesting that these two phenotypes belong to a continuous pathological spectrum. Nonmotor MSA is a noteworthy clinicopathological variant associated with a risk of unexpected death prior to fulfilling the current diagnostic criteria, which may arise from pathological involvement of the medullary serotonergic system.
- Although risk factors for a poor prognosis differ among reports, several natural history studies have found that severe autonomic failure and time from onset to concomitant presence of autonomic failure and motor involvement are commonly associated with rapid progression and survival [27-29]. It is urgent to establish a strategy for the early diagnosis of MSA during the isolated autonomic failure phase.
- Isolated parkinsonism or cerebellar ataxia phase in MSA
- Patients with MSA often show isolated parkinsonism or cerebellar ataxia. Particularly, the diagnosis of patients with a long duration of the isolated phase without red-flag signs is difficult. Petrovic et al. [30] reported that four of 135 autopsy cases showed MSA-P with slow progression and prolonged survival in the Queen Square Brain Bank. Although the mean survival time of patients with MSA is approximately 9 years, the disease duration of these four patients with MSA-P was 15 years or longer. Interestingly, the latency from the onset of parkinsonism to autonomic failure ranged from 9 to 14 years. The initial levodopa response was good in one, moderate in two, and poor in one patient. All patients developed dyskinesia after a mean duration of levodopa treatment of 4.3 years with generalized chorea. Red-flag signs were observed in all four patients. The mean latency of red-flag signs from onset was 9 years. Thus, the early diagnosis of these patients would not have been possible using the current consensus statement.
- The term idiopathic late onset cerebellar ataxia (ILOCA) was proposed in 1981 [31]. ILOCA is a slowly progressive, adult-onset ataxia and includes both ataxia and urinary dysfunction, abnormal reflexes, and dementia. Currently, ILOCA is considered to include MSA-C, spinocerebellar ataxias (SCAs), fragile X-associated tremor ataxia syndrome, Friedreich’s ataxia, autosomal recessive cerebellar ataxia type 1, and other novel genetic disorders. Thus, it is very important to distinguish early in the disease course between patients with MSA-C who show isolated cerebellar ataxia and those with other causes of ILOCA [32]. Gilman et al. [33] reported that the median time from isolated cerebellar ataxia (ILOCA phenotype) to the combination of cerebellar ataxia and autonomic failure (MSA) was 4.5 years.
- Figure 3 summarize the many faces of MSA. To diagnose patients with MSA who show isolated parkinsonism or cerebellar ataxia during the early course of the illness, other specific imaging modalities or biomarkers will be necessary.
MONOSYSTEM ATROPHY
- Dementia and hallucination in MSA
- Dementia based on the DSM-IV is a nonsupporting feature of MSA. However, patients with MSA frequently have fronto-executive dysfunction and may show impairment in memory and visuospatial function [34]. Imaging and neuropathological findings support the concept that striatofrontal deafferentation, cortical degeneration, and cerebellar pathology could be associated with cognitive impairments in MSA.
- A recent multicenter study using gray- and white-matter voxel-based morphometry and fully automated subcortical segmentation showed that only focal volume reduction in the left dorsolateral prefrontal cortex was observed in patients with MSA with cognitive decline compared to those with normal cognition, suggesting only a marginal contribution of cortical pathology to cognitive deficits [35]. Previously, we reported that brain SPECT showed a significant correlation between neuropsychological impairment and decreased perfusion in the prefrontal cortex in patients with MSA-P [36]. 18F-FDG brain PET showed that glucose metabolism in the striatum was the most powerful determinant of glucose metabolism in the frontal cortex in MSA-P [37]. Deafferentation due to focal degeneration of striatofrontal circuits may play an important role in frontal-executive dysfunction in some patients with MSA-P. However, it remains undetermined whether putamen-or white matter-tract involvement correlates with deafferentation and cognitive decline in MSA-P.
- In contrast, longitudinal studies have shown that progressive cerebral atrophy could occur in patients with MSA [38,39]. Kitayama et al. [40] reported that 10 of 58 patients with MSA (17%) had dementia fulfilling the DSM-IV criteria. More recently, autopsyconfirmed MSA cases that presented a clinical disease type of frontotemporal dementia (one with behavioral variant frontotemporal dementia, one with progressive nonfluent aphasia, and two with corticobasal syndrome) were reported [41]. These results also support the view that cortical changes may influence cognitive decline in MSA.
- Patients with MSA-C could also show similar cognitive decline compared to patients with MSA-P [34], but the pathogenic mechanism of neural correlates causing the cognitive impairments in MSA-C remains undetermined. It is well known that the greater part of the human cerebellum is associated with cerebral networks involved in cognition [42]. Since the frontal cortex and the cerebellar networks are composed of polysynaptic circuits, analysis of functional imaging such as of resting-state networks could provide insight into subcortical cognitive decline in MSA-C.
- Hallucinations not induced by drugs are also nonsupporting features of MSA. According to the results provided by the European Multiple System Atrophy registry, the frequency of patients experiencing hallucinations was only 5.5% [43]. A retrospective autopsy study reported that 9% of patients with MSA had hallucinations [44]. More recently, Koga et al. [13] demonstrated that visual hallucinations were observed in 10 of 79 (13%) patients with autopsy-confirmed MSA. The Neuropsychiatric Inventory revealed that 15% of patients with MSA had hallucinations [45]. Since these reports did not include detailed information about the use of dopaminergic medication, it is difficult to speculate on the pathophysiology of the hallucinations. Particularly, the influence of concomitant Lewy body pathology [46] should be considered since Lewy body-spectrum pathology was observed in 22.7% of patients with MSA [47].
- Other atypical symptoms in MSA
- Patients with MSA may show vertical gaze palsy, which is a characteristic finding of PSP. Koga et al. [13] reported that 17.6% of patients with autopsy-confirmed MSA presented vertical gaze palsy. However, the detailed clinical characteristics and pathological background of vertical gaze palsy in MSA have not been elucidated, since PSP, particularly PSP with a predominant cerebellar ataxia type, shows a combination of cerebellar ataxia and vertical gaze palsy. Age at onset, early falls, and no dysautonomia can increase the accuracy of the diagnosis of PSP-C [48].
ATYPICAL SYMPTOMS IN MSA
- Toward the development of diseasemodifying therapy or symptomatic interventions for MSA
- The etiology and pathogenesis of MSA remains unresolved but understanding of the intrinsic mechanism has steadily improved. Relocation of phosphoprotein-25α (p25α), which is a myelin stabilizing protein, from the myelin sheath to the oligodendroglial cell soma followed by formation of cytoplasmic p25α inclusions has been found to be an early event in patients with MSA [49]. Although the presence of α-syn in oligodendrocytes remains under debate, aberrant α-syn mRNA expression and α-syn prion-like propagation are considered to be closely associated with disease onset and progression [50]. Decreased glial cell line-derived neurotrophic factor expression, autophagy disruption, mitochondrial failure, and neuroinflammation can also play an important role in the pathogenesis of MSA [51]. Although MSA is reportedly a sporadic disease, rarely, patterns of familial aggregation following autosomal dominant and autosomal recessive inheritance have been reported, which supports the hypothesis of a genetic contribution [52]. Tsuji et al. [53] demonstrated that functionally impaired variants of COQ2 were associated with an increased risk of MSA in multiplex families and patients with sporadic disease. A recent meta-analysis reported an association of the COQ2 V393A variant with an increased risk of MSA in patients of East Asian ancestry [54].
- Clinical trials on disease-modifying therapy (DMT) have been conducted. A randomized, double-blind, placebo-controlled trial of autologous mesenchymal stem cells (MSCs) administered through the carotid artery revealed a significantly slower progression of the Unified Multiple System Atrophy Rating Scale (UMSARS) scores compared to placebo [55]. Although there were several limitations including a small number of patients, a single-center design, and increased signal abnormalities on brain MRI indicating small strokes, this study provided encouragement for researchers to conduct additional clinical trials. Consequently, a phase 1 study is underway at the Mayo Clinic in Rochester to evaluate the safety and tolerability of intrathecal injection of autologous MSCs in a dose-escalation study with patients with MSA (NCT02315027). The results of studies on myeloperoxidase inhibitors for ameliorating microglial activation (NCT02388295) and on two vaccines (PD01A and PD03A) against α-syn (NCT02270489) are also expected.
- For the successful development of DMT or symptomatic interventions for MSA, we need early diagnostic criteria and clinically definitive categorization. However, as we discussed in the previous section, early diagnosis of MSA can still be challenging based exclusively on clinical findings because patients with MSA can show isolated parkinsonism, cerebellar ataxia, and autonomic failure, particularly during the early course of the illness, as well as other atypical findings including cognitive impairment and vertical gaze palsy (Figure 4). Thus, it is crucial to establish well-validated biomarkers for a very early and clinically definitive diagnosis.
- Diagnostic challenges for isolated autonomic failure phase
- According to a United States prospective cohort study on PAF [19], the presence of a probable rapid-eye movement sleep behavior disorder is strongly correlated with the development of DLB, PD, or MSA. Patients whose diagnosis converted from PAF to MSA had a younger age at onset, severe bladder/bowel dysfunction, preserved olfactory function, and a cardiac chronotropic response to tilt more than 10 beats per minute. Patients with PAF that converted to DLB or PD showed impaired olfactory function, a lesser chronotropic response to tilt, and a longer duration of illness. The patients with retained PAF had very low plasma noradrenaline (NA) levels, no rapid eye movement (REM) sleep behavior disorder (RBD), preserved olfactory function, and a slow resting heart rate. The combination of olfactory function and plasma NA levels successfully classified PD/DLB (impaired olfaction, tended to have lower NA levels), PAF (normal olfaction and lower NA levels), and MSA (normal olfaction and tended to have plasma NA levels that were not low). Thus, the combination of age at onset of autonomic failure, bladder/bowel function, olfactory function, RBD, plasma NA levels, and a cardiac chronotropic response to tilt may serve as a good indicator for predicting conversion from PAF to MSA.
- Kikuchi et al. [56] reported that an olfactory function test can be valuable as a clinical instrument for differentiating PD from MSA in the early stages with high specificity. Low plasma NA levels correspond to postganglionic involvement in PD, DLB, and PAF [57]. If available, 123I-meta-iodobenzylguanidine (MIBG) cardiac scintigraphy may provide additional information for differentiating MSA from PD, DLB, and PAF [58,59]. In general, PD, DLB, and PAF show predominantly postganglionic involvement, but MSA shows preganglionic involvement [19]. However, to varying degrees, both preganglionic and postganglionic involvements can be observed in MSA [60]. Nagayama et al. [61] found that there was significant longitudinal decreased cardiac MIBG accumulation in patients with MSA. Patients with MSA may have Lewy body pathology including cardiac sympathetic nervous system involvement [62,63]. Thus, we should consider the possibility of a complex pathology and differences in the spread of the autonomic involvement in MSA.
- Although PAF is a rare condition, further studies are expected to prove the usefulness of a composite score for these indices, to compare them with the autopsy results and to develop additional semi- or quantitative biomarkers that will improve the objective efficacy of DMT.
- Diagnostic challenge for an isolated parkinsonism phase
- Conventional MRI (cMRI) is a reliable and widely available test for differentiating MSA from PD, DLB, and PSP. Based on a detailed review of brain MRI as a diagnostic tool for MSA, several characteristic MRI findings have been incorporated in the current diagnostic criteria as supportive of ‘possible MSA.’ [10] However, the utility of these findings has not been investigated for patients with MSA during the early stages, when the clinical diagnosis remains uncertain.
- Recently, Mestre et al. [64] reported that 30% of patients with MSA-P showed characteristic MRI signs preceding the clinical diagnosis. T2 posterior putaminal atrophy affected 91.7% of the patients (n = 11/12), T2 slit-like putaminal hyperintensity 50% (n = 6/12), and hypointensity and/ or definite atrophy of the posterior putamen on T2 imaging 66.7% (6/9), which were suggested as characteristic abnormal MRI findings for MSA. These results support the view that the diagnostic criteria should be reconsidered based on cMRI findings.
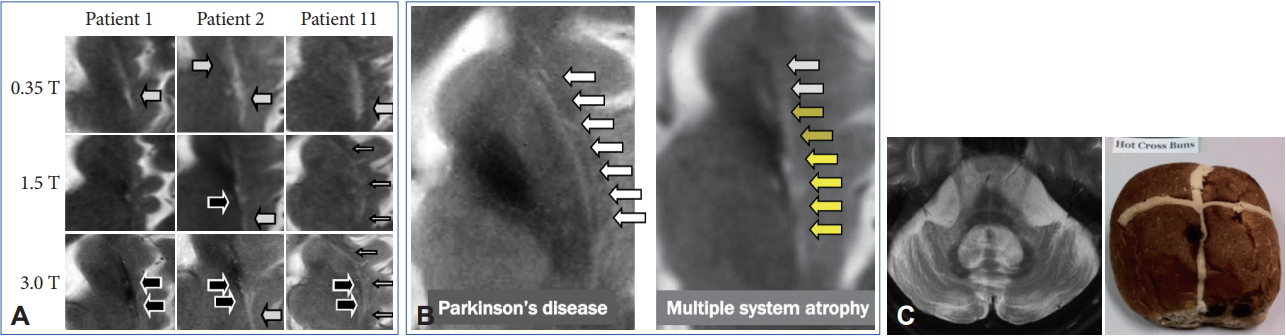
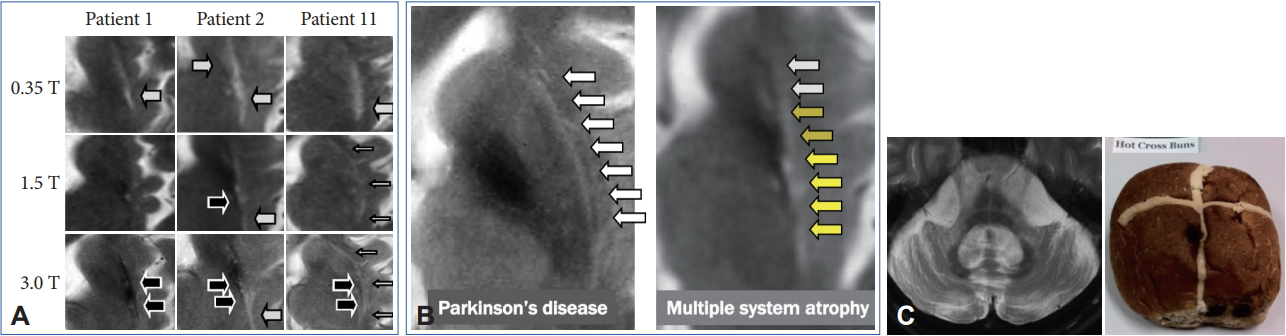
- However, these cMRI findings may also be observed not only in patients with PD, PSP, SCA 17, and adult GM1 gangliosidosis but also in control subjects [65-67]. In addition, putaminal MRI findings may change among different magnetic-field strengths (Figure 5A) [68,69]. A T2 hyperintense putaminal rim without atrophic changes at 3 T may be non-specifically observed in patients with PD or healthy controls (Figure 5B) but the configuration of PD or controls and MSA can be different. Finally, we should also consider that approximately 60% of patients with MSA-P had normal cMRI findings within 2 years of disease onset [14].
- 123I-MIBG cardiac scintigraphy can also be useful for differentiating PD from other neurodegenerative parkinsonism conditions such as MSA and PSP. However, it is difficult to differentiate MSA from PSP and corticobasal degeneration (CBD) since patients with MSA, PSP, and CBD will show preserved MIBG accumulation [58]. In addition, there are no data regarding the results of MIBG cardiac scintigraphy findings in patients with early MSA who did not fulfill the diagnostic criteria.
- Diagnostic challenges for isolated cerebellar ataxia phase
- Pontine signal alterations called a hot cross bun (HCB) sign and T2 hyperintensity in the bilateral middle cerebellar peduncle (MCP) are characteristic MRI findings in patients with MSA (Figure 5C). Interestingly, SCA 2, SCA 3, SCA 7, and SCA 8 may also show an HCB sign [70]. T2 hyperintensity in the MCP is also observed in patients with fragile X associated tremor/ataxia syndrome, Wilson’s disease, liver cirrhosis, multiple sclerosis, stroke and others [71-73]. Kim et al. [74] demonstrated that SCA 1, 2, 3, 6, 17, or dentatorubral-pallidoluysian atrophy was confirmed in 22 of 302 (7.3%) of clinically diagnosed MSA cases. Thus, we should consider both MSA and SCAs when patients show a combination of an HCB sign and T2 hyperintensity in the MCP.
- However, previous studies have not given due consideration to disease duration from onset to MRI. In general, the progression rate is significantly rapid in MSA compared to SCAs [75]. Importantly, more than 60% of patients with MSA-C had an HCB sign within 2 years from the onset of ataxia [14]. In contrast, patients with SCAs with an HCB sign showed significantly longer disease duration compared to MSA-C [76,77] although available data are limited. Careful neurological evaluation to exclude nonneurodegenerative diseases will be necessary, but an early HCB sign may strongly support the diagnosis of MSA.
WHY DO WE NEED NEW DIAGNOSTIC CRITERIA FOR MSA?
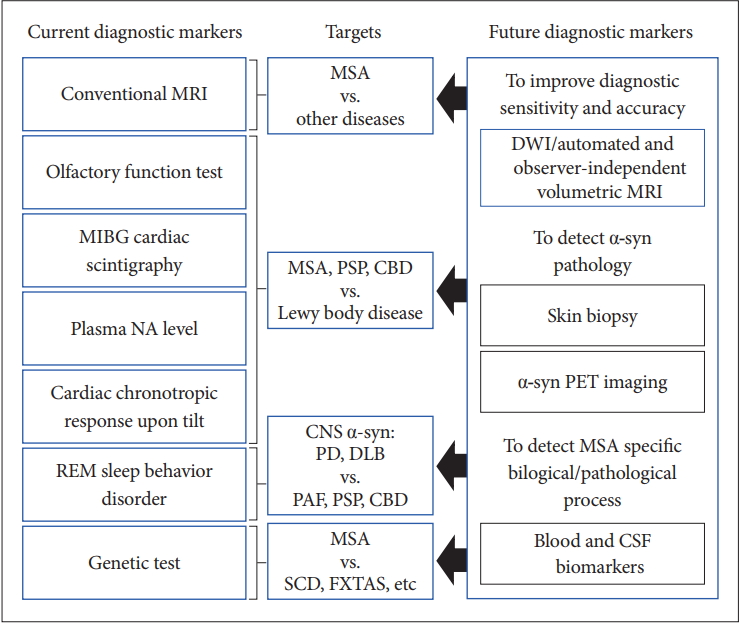
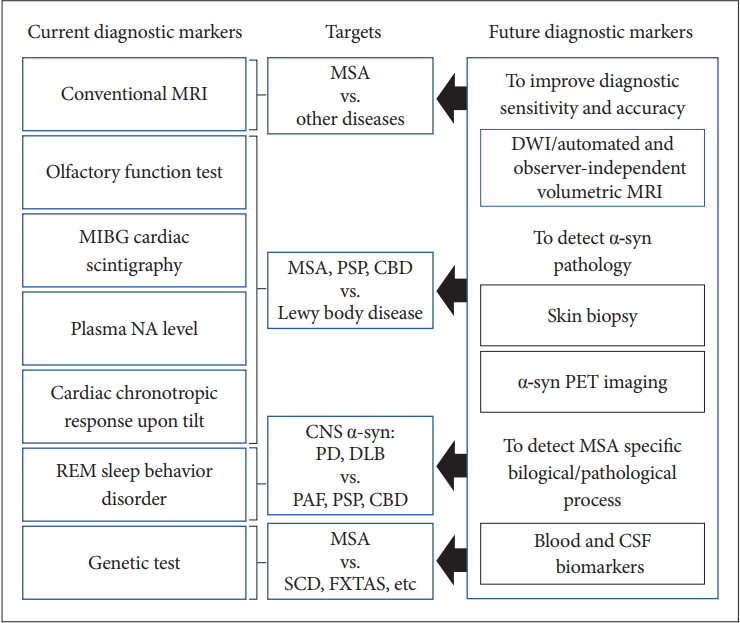
- Currently, conventional MRI, olfactory test, 123IMIBG cardiac scintigraphy, plasma NA level, cardiac chronotropic response upon tilt, REM sleep behavior disorder, and genetic tests and so on are available for the differential diagnosis of MSA from other diseases in clinical practice. However, there is no established test for the early and clinically definitive diagnosis that can satisfy the criteria of objectivity, semi- or quantitative measurement, and uncomplicated and worldwide availability.
- MRI
- Diffusion-weighted MRI (DWI) measures the random Brownian motion of water molecules within a voxel. Patients with MSA-P show increased putaminal diffusivity compared to those with PD [78-80]. A meta-analysis on putaminal diffusivity showed a sensitivity of 90% [95% confidence interval (CI): 76.7–95.8%] and specificity of 93% (95% CI: 80.0–97.7%) for discriminating MSA-P from PD [81]. More recently, Péran et al. [82] demonstrated that multimodal MRI, including DWI, is able to discriminate patients with PD from those with MSA with high accuracy. Further prospective multicenter studies with standardized regions of interest and imaging protocols across institutions or different MRI equipment vendors of DWI will be needed, particularly of cases in the early disease stages prior to fulfilling the diagnostic criteria.
- Voxel-based morphometry analysis is a wholebrain, unbiased, objective technique. Recent advances in imaging algorithms have enabled automatic compartmentalization of the brain into multiple parcellations and provide for quantification of gray and white matter volumes in these regions on an individual basis [83]. Scherfler et al. [84] demonstrated that the diagnostic accuracy for PD vs. MSA-P or PSP was 97.4% using the midbrain and putaminal volumes as well as cerebellar gray matter volume calculated by automated and observer-independent volumetric MRI analysis, although the diagnostic accuracy based on validated clinical consensus criteria was 62.9%. Compared to DWI, multicenter studies have widely used volumetry. Individual volumetric T1-weighted MRI can be a good candidate for a novel diagnostic marker for novel diagnostic criteria for MSA. We should confirm the usefulness of this procedure for the diagnosis of patients with isolated autonomic failure and in the cerebellar ataxia phase.
- Visualization of α-syn
- Biopsy and PET are two modalities used to detect α-syn accumulation in MSA. Doppler et al. [85] demonstrated that phospho-α-syn (p-α-syn) in dermal nerve fibers was found in 67% of patients with MSA and PD, but not in those with tauopathy or controls when analyzing 15 consecutive sections. Analyzing serial sections increased the sensitivity to 75% and 73%, respectively. Interestingly, p-α-syn clustered in autonomic fibers in PD but mainly in unmyelinated somatosensory fibers in MSA.
- BF-227 can bind aggregated amyloid beta protein and p-α-syn. The PET data of BF 227 demonstrated high distribution volumes in the subcortical white matter, putamen and posterior cingulate cortex, globus pallidus, primary motor cortex and anterior cingulate cortex, and substantia nigra in MSA in accordance with GCI-rich brain areas compared to the normal controls. A pathological study showed that BF-227 histofluorescence was observed in most of the GCIs [86]. However, Verdurand et al. [87] reported that 18F-BF-227 did not bind to GCIs of MSA brain tissue using state-of-the-art autoradiography.
- More recently, Koga et al. [88] demonstrated that the tau tracer 11C-PBB3 showed significant autoradiographic binding to the striatopallidal fibers in two MSA cases with high densities of GCIs. One MSA case showed increased binding of PBB3 in the frontal lobe, globus pallidus, midbrain, parietal lobe, putamen, temporal lobe, substantia nigra, thalamus, and ventral striatum [89].
- Although there are several candidate PET radiotracers for imaging aggregated α-syn, they are not easy to develop. The amount of insoluble α-syn protein in the MSA brain is 10-fold or lower than the amount of Aβ in the Alzheimer’s disease brain. The tracer must readily pass through the glial cell membrane to access the binding site [90]. There is still no known radioligand that selectively binds to α-syn with high affinity.
- Other candidate biomarkers
- MSA, particularly MSA-P, showed significantly decreased peripapillary retinal nerve fiber layer thickness in the inferior and inferotemporal sectors and significant perifoveal thinning in the superior outer sector compared to healthy controls. Retinal thinning correlated with the UMSARS score and the Geriatric Depression Scale [91].
- A number of studies have searched for candidate biomarkers using blood and cerebrospinal fluid, including neurofilament light chain, catecholamine metabolite, coenzyme Q10 micro RNA, and diseaserelated proteins such as total α-syn, DJ-1, amyloid beta and total tau, but there are still no reliable biomarkers for the diagnosis of MSA [92-99].
- Figure 6 summarizes the current and future diagnostic biomarkers of MSA.
DEVELOPMENT OF NOVEL DIAGNOSTIC BIOMARKERS
- The several trials assessing potential DMT failed to reach their primary efficacy endpoint, although preclinical disease-model studies have shown positive findings [100-103]. As we discussed in this review, probable MSA can only be diagnosed at a substantially advanced stage. Early diagnostic criteria and clinically definitive categorization are needed for the success of DMT or symptomatic interventions for MSA. To develop early diagnostic criteria, prospective image-omics cohort studies focusing on patients with MSA who show isolated autonomic failure or motor involvement will be necessary. Since the MSA phenotype could differ between western and eastern countries (MSA-C vs. MSA-P, proportion of the positive COQ2 mutations, etc.) [104,105], international collaborative studies will be helpful. To establish clinically definitive criteria, the development of a disease-specific marker is urgently needed because patients with MSA may show atypical symptoms during the course of the illness. PD, PSP, DLB, PAF, and other genetic, and immunologic conditions can mimic MSA [106]. Molecular PET imaging and biopsies for detecting α-syn are promising, but we need more specific radioligands and multicenter results. On the other hand, individually automated and observer-independent volumetric MRI analysis seem to be attractive because it will satisfy the requirements for objectivity, semi- or quantitative measurements, and uncomplicated and worldwide availability for universal use. Further prospective multicenter studies will be necessary to incorporate this technique into future diagnostic criteria. Finally, the combination of multimodal markers, including functional and molecular imaging, as well as cerebrospinal fluid and plasma biomarkers, will be helpful for both establishing early diagnostic criteria and for clinically definitive categorization.
CONCLUSIONS
- This work was supported by Grants-in-Aid from the Research Committee of Central Nervous System Degenerative Diseases by the Ministry of Health, Labour, and Welfare and from the Integrated Research on Neuropsychiatric Disorders project carried out under the Strategic Research for Brain Sciences by the Ministry of Education, Culture, Sports, Science, and Technology of Japan. This work was also supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan (grant number 80569781), and a Grant-in-Aid for Scientific Research on Innovative Areas (Brain Protein Aging and Dementia Control) (26117002) from MEXT.
Acknowledgments

| Supporting features (Red flag signs) [10] |
| Orofacial dystonia |
| Disproportionate antecollis |
| Camptocormia and/or Pisa syndrome |
| Contractures of hands or feet |
| Inspiratory sighs |
| Severe dysphonia |
| Severe dysarthria |
| New or increased snoring |
| Cold hands and feet |
| Pathologic laughter or crying |
| Jerky, myoclonic postural/action tremor |
| Nonsupporting features [10] |
| Classic pill-rolling rest tremor |
| Clinically significant neuropathy |
| Hallucination not induced by drug |
| Onset after age 75 years |
| Family history of ataxia or parkinsonism |
| Dementia (on DSM-IV) |
| White matter lesions suggesting multiple sclerosis |
- 1. DeÂjerine J, Thomas AA. L’atrophie olivo-ponto-ceÂreÂbelleuse. Nouv lconogr SalpeÃtr 1900;13:330–70.
- 2. Shy GM, Drager GA. A neurological syndrome associated with orthostatic hypotension: a clinical-pathologic study. Arch Neurol 1960;2:511–527.ArticlePubMed
- 3. Adams RD, van Bogaert L, van der Eecken H. Striato-nigral degeneration. J Neuropathol Exp Neurolol 1964;23:584–608.
- 4. Graham JG, Oppenheimer DR. Orthostatic hypotension and nicotine sensitivity in a case of multiple system atrophy. J Neurol Neurosurg Psychiatry 1969;32:28–34.ArticlePubMedPMC
- 5. Takahashi A, Takagi S, Yamamoto K, Yamada T, Ando K. [Shy-Drager syndrome. Its correlation with olivo-ponto-cerebellar atrophy]. Rinsho Shinkeigaku 1969;9:121–129.
- 6. Papp MI, Kahn JE, Lantos PL. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). J Neurol Sci 1989;94:79–100.ArticlePubMed
- 7. Nakazato Y, Yamazaki H, Hirato J, Ishida Y, Yamaguchi H. Oligodendroglial microtubular tangles in olivopontocerebellar atrophy. J Neuropathol Exp Neurol 1990;49:521–530.ArticlePubMedPDF
- 8. Wakabayashi K, Yoshimoto M, Tsuji S, Takahashi H. Alphasynuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci Lett 1998;249:180–182.ArticlePubMed
- 9. Gilman S, Low PA, Quinn N, Albanese A, Ben-Shlomo Y, Fowler CJ, et al. Consensus statement on the diagnosis of multiple system atrophy. J Neurol Sci 1999;163:94–98.ArticlePubMed
- 10. Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670–676.ArticlePubMedPMC
- 11. Köllensperger M, Geser F, Seppi K, Stampfer-Kountchev M, Sawires M, Scherfler C, et al. Red flags for multiple system atrophy. Mov Disord 2008;23:1093–1099.ArticlePubMed
- 12. Osaki Y, Ben-Shlomo Y, Lees AJ, Wenning GK, Quinn NP. A validation exercise on the new consensus criteria for multiple system atrophy. Mov Disord 2009;24:2272–2276.ArticlePubMed
- 13. Koga S, Aoki N, Uitti RJ, van Gerpen JA, Cheshire WP, Josephs KA, et al. When DLB, PD, and PSP masquerade as MSA: an autopsy study of 134 patients. Neurology 2015;85:404–412.ArticlePubMedPMC
- 14. Watanabe H, Saito Y, Terao S, Ando T, Kachi T, Mukai E, et al. Progression and prognosis in multiple system atrophy: an analysis of 230 Japanese patients. Brain 2002;125(Pt 5):1070–1083.ArticlePubMedPDF
- 15. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinicopathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–184.ArticlePubMedPMC
- 16. Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord 2015;30:1591–1601.ArticlePubMed
- 17. Velseboer DC, de Haan RJ, Wieling W, Goldstein DS, de Bie RM. Prevalence of orthostatic hypotension in Parkinson’s disease: a systematic review and meta-analysis. Parkinsonism Relat Disord 2011;17:724–729.ArticlePubMedPMC
- 18. Thaisetthawatkul P, Boeve BF, Benarroch EE, Sandroni P, Ferman TJ, Petersen R, et al. Autonomic dysfunction in dementia with Lewy bodies. Neurology 2004;62:1804–1809.ArticlePubMed
- 19. Kaufmann H, Norcliffe-Kaufmann L, Palma JA, Biaggioni I, Low PA, Singer W, et al. Natural history of pure autonomic failure: a United States prospective cohort. Ann Neurol 2017;81:287–297.ArticlePubMedPMC
- 20. Gaig C, Iranzo A, Tolosa E, Vilaseca I, Rey MJ, Santamaria J. Pathological description of a non-motor variant of multiple system atrophy. J Neurol Neurosurg Psychiatry 2008;79:1399–1400.ArticlePubMed
- 21. Riku Y, Watanabe H, Mimuro M, Iwasaki Y, Ito M, Katsuno M, et al. Non-motor multiple system atrophy associated with sudden death: pathological observations of autonomic nuclei. J Neurol 2017;264:2249–2257.ArticlePubMedPDF
- 22. Benarroch EE. The clinical approach to autonomic failure in neurological disorders. Nat Rev Neurol 2014;10:396–407.ArticlePubMedPDF
- 23. Cersosimo MG, Benarroch EE. Central control of autonomic function and involvement in neurodegenerative disorders. Handb Clin Neurol 2013;117:45–57.ArticlePubMed
- 24. Tada M, Kakita A, Toyoshima Y, Onodera O, Ozawa T, Morita T, et al. Depletion of medullary serotonergic neurons in patients with multiple system atrophy who succumbed to sudden death. Brain 2009;132(Pt 7):1810–1819.ArticlePubMedPDF
- 25. Figueroa JJ, Singer W, Parsaik A, Benarroch EE, Ahlskog JE, Fealey RD, et al. Multiple system atrophy: prognostic indicators of survival. Mov Disord 2014;29:1151–1157.ArticlePubMedPMC
- 26. Ling H, Asi YT, Petrovic IN, Ahmed Z, Prashanth LK, Hazrati LN, et al. Minimal change multiple system atrophy: an aggressive variant? Mov Disord 2015;30:960–967.ArticlePubMed
- 27. Wenning GK, Geser F, Krismer F, Seppi K, Duerr S, Boesch S, et al. The natural history of multiple system atrophy: a prospective European cohort study. Lancet Neurol 2013;12:264–274.ArticlePubMedPMC
- 28. Coon EA, Sletten DM, Suarez MD, Mandrekar JN, Ahlskog JE, Bower JH, et al. Clinical features and autonomic testing predict survival in multiple system atrophy. Brain 2015;138(Pt 12):3623–3631.ArticlePubMedPMCPDF
- 29. Low PA, Reich SG, Jankovic J, Shults CW, Stern MB, Novak P, et al. Natural history of multiple system atrophy in the USA: a prospective cohort study. Lancet Neurol 2015;14:710–719.ArticlePubMedPMC
- 30. Petrovic IN, Ling H, Asi Y, Ahmed Z, Kukkle PL, Hazrati LN, et al. Multiple system atrophy-parkinsonism with slow progression and prolonged survival: a diagnostic catch. Mov Disord 2012;27:1186–1190.ArticlePubMed
- 31. Harding AE. “Idiopathic” late onset cerebellar ataxia. A clinical and genetic study of 36 cases. J Neurol Sci 1981;51:259–271.ArticlePubMed
- 32. Lin DJ, Hermann KL, Schmahmann JD. Multiple system atrophy of the cerebellar type: clinical state of the art. Mov Disord 2014;29:294–304.ArticlePubMed
- 33. Gilman S, Little R, Johanns J, Heumann M, Kluin KJ, Junck L, et al. Evolution of sporadic olivopontocerebellar atrophy into multiple system atrophy. Neurology 2000;55:527–532.ArticlePubMed
- 34. Stankovic I, Krismer F, Jesic A, Antonini A, Benke T, Brown RG, et al. Cognitive impairment in multiple system atrophy: a position statement by the Neuropsychology Task Force of the MDS Multiple System Atrophy (MODIMSA) study group. Mov Disord 2014;29:857–867.ArticlePubMedPMC
- 35. Fiorenzato E, Weis L, Seppi K, Onofrj M, Cortelli P, Zanigni S, et al. Brain structural profile of multiple system atrophy patients with cognitive impairment. J Neural Transm (Vienna) 2017;124:293–302.ArticlePubMedPDF
- 36. Kawai Y, Suenaga M, Takeda A, Ito M, Watanabe H, Tanaka F, et al. Cognitive impairments in multiple system atrophy: MSA-C vs MSA-P. Neurology 2008;70(16 Pt 2):1390–1396.ArticlePubMed
- 37. Kim HW, Oh M, Oh JS, Oh SJ, Lee SJ, Chung SJ, et al. Striatofrontal Deafferentiation in MSA-P: Evaluation with [18F]FDG Brain PET. PLoS One 2017;12:e0169928.ArticlePubMedPMC
- 38. Horimoto Y, Aiba I, Yasuda T, Ohkawa Y, Katayama T, Yokokawa Y, et al. Cerebral atrophy in multiple system atrophy by MRI. J Neurol Sci 2000;173:109–112.ArticlePubMed
- 39. Konagaya M, Sakai M, Matsuoka Y, Konagaya Y, Hashizume Y. Multiple system atrophy with remarkable frontal lobe atrophy. Acta Neuropathol 1999;97:423–428.ArticlePubMedPDF
- 40. Kitayama M, Wada-Isoe K, Irizawa Y, Nakashima K. Assessment of dementia in patients with multiple system atrophy. Eur J Neurol 2009;16:589–594.ArticlePubMed
- 41. Aoki N, Boyer PJ, Lund C, Lin WL, Koga S, Ross OA, et al. Atypical multiple system atrophy is a new subtype of frontotemporal lobar degeneration: frontotemporal lobar degeneration associated with α-synuclein. Acta Neuropathol 2015;130:93–105.ArticlePubMedPMCPDF
- 42. Sokolov AA, Miall RC, Ivry RB. The cerebellum: adaptive prediction for movement and cognition. Trends Cogn Sci 2017;21:313–332.ArticlePubMedPMC
- 43. Köllensperger M, Geser F, Ndayisaba JP, Boesch S, Seppi K, Ostergaard K, et al. Presentation, diagnosis, and management of multiple system atrophy in Europe: final analysis of the European multiple system atrophy registry. Mov Disord 2010;25:2604–2612.ArticlePubMed
- 44. Williams DR, Lees AJ. Visual hallucinations in the diagnosis of idiopathic Parkinson’s disease: a retrospective autopsy study. Lancet Neurol 2005;4:605–610.ArticlePubMed
- 45. Ceponiene R, Edland SD, Reid TN, Al Rizaiza1 A, Litvan I. Neuropsychiatric symptoms and their impact on quality of life in multiple system atrophy. Cogent Psychol 2016;3:1131476.ArticlePDF
- 46. Cykowski MD, Coon EA, Powell SZ, Jenkins SM, Benarroch EE, Low PA, et al. Expanding the spectrum of neuronal pathology in multiple system atrophy. Brain 2015;138(Pt 8):2293–2309.ArticlePubMedPMCPDF
- 47. Jellinger KA. More frequent Lewy bodies but less frequent Alzheimer-type lesions in multiple system atrophy as compared to age-matched control brains. Acta Neuropathol 2007;114:299–303.ArticlePubMedPDF
- 48. Kanazawa M, Tada M, Onodera O, Takahashi H, Nishizawa M, Shimohata T. Early clinical features of patients with progressive supranuclear palsy with predominant cerebellar ataxia. Parkinsonism Relat Disord 2013;19:1149–1151.ArticlePubMed
- 49. Song YJ, Lundvig DM, Huang Y, Gai WP, Blumbergs PC, Højrup P, et al. p25alpha relocalizes in oligodendroglia from myelin to cytoplasmic inclusions in multiple system atrophy. Am J Pathol 2007;171:1291–1303.ArticlePubMedPMC
- 50. Woerman AL, Stöhr J, Aoyagi A, Rampersaud R, Krejciova Z, Watts JC, et al. Propagation of prions causing synucleinopathies in cultured cells. Proc Natl Acad Sci U S A 2015;112:E4949–E4958.ArticlePubMedPMC
- 51. Krismer F, Wenning GK. Multiple system atrophy: insights into a rare and debilitating movement disorder. Nat Rev Neurol 2017;13:232–243.ArticlePubMedPDF
- 52. Hara K, Momose Y, Tokiguchi S, Shimohata M, Terajima K, Onodera O, et al. Multiplex families with multiple system atrophy. Arch Neurol 2007;64:545–551.ArticlePubMed
- 53. Multiple-System Atrophy Research Collaboration. Mutations in COQ2 in familial and sporadic multiple-system atrophy. N Engl J Med 2013;369:233–244.ArticlePubMed
- 54. Yang X, Xi J, Zhao Q, Jia H, An R, Liu Z, et al. Association of the COQ2 V393A variant with Parkinson’s disease: a casecontrol study and meta-analysis. PLoS One 2015;10:e0130970.ArticlePubMedPMC
- 55. Lee PH, Lee JE, Kim HS, Song SK, Lee HS, Nam HS, et al. A randomized trial of mesenchymal stem cells in multiple system atrophy. Ann Neurol 2012;72:32–40.ArticlePubMed
- 56. Kikuchi A, Baba T, Hasegawa T, Sugeno N, Konno M, Takeda A. Differentiating Parkinson’s disease from multiple system atrophy by [123I] meta-iodobenzylguanidine myocardial scintigraphy and olfactory test. Parkinsonism Relat Disord 2011;17:698–700.ArticlePubMed
- 57. Jordan J, Shibao C, Biaggioni I. Multiple system atrophy: using clinical pharmacology to reveal pathophysiology. Clin Auton Res 2015;25:53–59.ArticlePubMedPMCPDF
- 58. Orimo S, Yogo M, Nakamura T, Suzuki M, Watanabe H. (123)I-meta-iodobenzylguanidine (MIBG) cardiac scintigraphy in α-synucleinopathies. Ageing Res Rev 2016;30:122–133.ArticlePubMed
- 59. Mabuchi N, Hirayama M, Koike Y, Watanabe H, Ito H, Kobayashi R, et al. Progression and prognosis in pure autonomic failure (PAF): comparison with multiple system atrophy. J Neurol Neurosurg Psychiatry 2005;76:947–952.ArticlePubMedPMC
- 60. Coon EA, Fealey RD, Sletten DM, Mandrekar JN, Benarroch EE, Sandroni P, et al. Anhidrosis in multiple system atrophy involves pre- and postganglionic sudomotor dysfunction. Mov Disord 2017;32:397–404.ArticlePubMed
- 61. Nagayama H, Ueda M, Yamazaki M, Nishiyama Y, Hamamoto M, Katayama Y. Abnormal cardiac [(123)I]-meta-iodobenzylguanidine uptake in multiple system atrophy. Mov Disord 2010;25:1744–1747.ArticlePubMed
- 62. Nagayama H, Yamazaki M, Ueda M, Nishiyama Y, Hamamoto M, Katayama Y, et al. Low myocardial MIBG uptake in multiple system atrophy with incidental Lewy body pathology: an autopsy case report. Mov Disord 2008;23:1055–1057.Article
- 63. Orimo S, Kanazawa T, Nakamura A, Uchihara T, Mori F, Kakita A, et al. Degeneration of cardiac sympathetic nerve can occur in multiple system atrophy. Acta Neuropathol 2007;113:81–86.ArticlePubMedPDF
- 64. Mestre TA, Gupta A, Lang AE. MRI signs of multiple system atrophy preceding the clinical diagnosis: the case for an imaging-supported probable MSA diagnostic category. J Neurol Neurosurg Psychiatry 2016;87:443–444.ArticlePubMed
- 65. Massey LA, Micallef C, Paviour DC, O’Sullivan SS, Ling H, Williams DR, et al. Conventional magnetic resonance imaging in confirmed progressive supranuclear palsy and multiple system atrophy. Mov Disord 2012;27:1754–1762.ArticlePubMed
- 66. Lin IS, Wu RM, Lee-Chen GJ, Shan DE, Gwinn-Hardy K. The SCA17 phenotype can include features of MSA-C, PSP and cognitive impairment. Parkinsonism Relat Disord 2007;13:246–249.ArticlePubMed
- 67. Muthane U, Chickabasaviah Y, Kaneski C, Shankar SK, Narayanappa G, Christopher R, et al. Clinical features of adult GM1 gangliosidosis: report of three Indian patients and review of 40 cases. Mov Disord 2004;19:1334–1341.ArticlePubMed
- 68. Watanabe H, Ito M, Fukatsu H, Senda J, Atsuta N, Kaga T, et al. Putaminal magnetic resonance imaging features at various magnetic field strengths in multiple system atrophy. Mov Disord 2010;25:1916–1923.ArticlePubMed
- 69. Watanabe H, Fukatsu H, Hishikawa N, Hashizume Y, Sobue G. Field strengths and sequences influence putaminal MRI findings in multiple system atrophy. Neurology 2004;62:671.ArticlePubMed
- 70. Lee YC, Liu CS, Wu HM, Wang PS, Chang MH, Soong BW. The ‘hot cross bun’ sign in the patients with spinocerebellar ataxia. Eur J Neurol 2009;16:513–516.ArticlePubMed
- 71. Okamoto K, Tokiguchi S, Furusawa T, Ishikawa K, Quardery AF, Shinbo S, et al. MR features of diseases involving bilateral middle cerebellar peduncles. AJNR Am J Neuroradiol 2003;24:1946–1954.PubMedPMC
- 72. Uchino A, Sawada A, Takase Y, Kudo S. Symmetrical lesions of the middle cerebellar peduncle: MR imaging and differential diagnosis. Magn Reson Med Sci 2004;3:133–140.ArticlePubMed
- 73. Yata S, Ogawa T, Sugihara S, Matsusue E, Fujii S, Kinoshita T. HTLV-I carrier with unusual brain MR imaging findings. Neuroradiology 2004;46:755–758.ArticlePubMedPDF
- 74. Kim HJ, Jeon BS, Shin J, Lee WW, Park H, Jung YJ, et al. Should genetic testing for SCAs be included in the diagnostic workup for MSA? Neurology 2014;83:1733–1738.ArticlePubMed
- 75. Klockgether T, Lüdtke R, Kramer B, Abele M, Bürk K, Schöls L, et al. The natural history of degenerative ataxia: a retrospective study in 466 patients. Brain 1998;121(Pt 4):589–600.ArticlePubMedPDF
- 76. Murata Y, Yamaguchi S, Kawakami H, Imon Y, Maruyama H, Sakai T, et al. Characteristic magnetic resonance imaging findings in Machado-Joseph disease. Arch Neurol 1998;55:33–37.ArticlePubMed
- 77. Higashi M, Ozaki K, Hattori T, Ishii T, Soga K, Sato N, et al. A diagnostic decision tree for adult cerebellar ataxia based on pontine magnetic resonance imaging. J Neurol Sci 2018;387:187–195.ArticlePubMed
- 78. Schocke MF, Seppi K, Esterhammer R, Kremser C, Jaschke W, Poewe W, et al. Diffusion-weighted MRI differentiates the Parkinson variant of multiple system atrophy from PD. Neurology 2002;58:575–580.ArticlePubMed
- 79. Seppi K, Schocke MF, Mair KJ, Esterhammer R, Scherfler C, Geser F, et al. Progression of putaminal degeneration in multiple system atrophy: a serial diffusion MR study. Neuroimage 2006;31:240–245.ArticlePubMed
- 80. Ito M, Watanabe H, Kawai Y, Atsuta N, Tanaka F, Naganawa S, et al. Usefulness of combined fractional anisotropy and apparent diffusion coefficient values for detection of involvement in multiple system atrophy. J Neurol Neurosurg Psychiatry 2007;78:722–728.ArticlePubMedPMC
- 81. Bajaj S, Krismer F, Palma JA, Wenning GK, Kaufmann H, Poewe W, et al. Diffusion-weighted MRI distinguishes Parkinson disease from the parkinsonian variant of multiple system atrophy: A systematic review and meta-analysis. PLoS One 2017;12:e0189897. ArticlePubMedPMC
- 82. Péran P, Barbagallo G, Nemmi F, Sierra M, Galitzky M, Traon AP, et al. MRI supervised and unsupervised classification of Parkinson’s disease and multiple system atrophy. Mov Disord 2018;33:600–608.ArticlePubMed
- 83. Fischl B, Dale AM. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci U S A 2000;97:11050–11055.ArticlePubMedPMC
- 84. Scherfler C, Göbel G, Müller C, Nocker M, Wenning GK, Schocke M, et al. Diagnostic potential of automated subcortical volume segmentation in atypical parkinsonism. Neurology 2016;86:1242–1249.ArticlePubMed
- 85. Doppler K, Weis J, Karl K, Ebert S, Ebentheuer J, Trenkwalder C, et al. Distinctive distribution of phospho-alphasynuclein in dermal nerves in multiple system atrophy. Mov Disord 2015;30:1688–1692.ArticlePubMed
- 86. Kikuchi A, Takeda A, Okamura N, Tashiro M, Hasegawa T, Furumoto S, et al. In vivo visualization of alpha-synuclein deposition by carbon-11-labelled 2-[2-(2-dimethylaminothiazol-5-yl)ethenyl]-6-[2-(fluoro)ethoxy]benzoxazole positron emission tomography in multiple system atrophy. Brain 2010;133(Pt 6):1772–1178.ArticlePubMedPDF
- 87. Verdurand M, Levigoureux E, Lancelot S, Zeinyeh W, Billard T, Quadrio I, et al. Amyloid-beta radiotracer [18F]BF-227 does not bind to cytoplasmic glial inclusions of postmortem multiple system atrophy brain tissue. Contrast Media Mol Imaging 2018;2018:9165458.ArticlePubMedPMCPDF
- 88. Koga S, Ono M, Sahara N, Higuchi M, Dickson DW. Fluorescence and autoradiographic evaluation of tau PET ligand PBB3 to α-synuclein pathology. Mov Disord 2017;32:884–892.ArticlePubMedPMC
- 89. Perez-Soriano A, Arena JE, Dinelle K, Miao Q, McKenzie J, Neilson N, et al. PBB3 imaging in Parkinsonian disorders: Evidence for binding to tau and other proteins. Mov Disord 2017;32:1016–1024.ArticlePubMed
- 90. Mathis CA, Lopresti BJ, Ikonomovic MD, Klunk WE. Small-molecule PET tracers for imaging proteinopathies. Semin Nucl Med 2017;47:553–575.ArticlePubMedPMC
- 91. Ahn J, Lee JY, Kim TW. Retinal thinning correlates with clinical severity in multiple system atrophy. J Neurol 2016;263:2039–2047.ArticlePubMedPDF
- 92. Marques TM, Kuiperij HB, Bruinsma IB, van Rumund A, Aerts MB, Esselink RAJ, et al. MicroRNAs in cerebrospinal fluid as potential biomarkers for Parkinson’s disease and multiple system atrophy. Mol Neurobiol 2017;54:7736–7745.ArticlePubMedPDF
- 93. Laurens B, Constantinescu R, Freeman R, Gerhard A, Jellinger K, Jeromin A, et al. Fluid biomarkers in multiple system atrophy: a review of the MSA Biomarker Initiative. Neurobiol Dis 2015;80:29–41.ArticlePubMed
- 94. Chen D, Wei X, Zou J, Wang R, Liu X, Xu X, et al. Contradirectional expression of serum homocysteine and uric acid as important biomarkers of multiple system atrophy severity: a cross-sectional study. Front Cell Neurosci 2015;9:247.ArticlePubMedPMC
- 95. Zhou L, Jiang Y, Zhu C, Ma L, Huang Q, Chen X. Oxidative stress and environmental exposures are associated with multiple system atrophy in Chinese patients. Can J Neurol Sci 2016;43:703–709.ArticlePubMed
- 96. Guo Y, Zhuang XD, Xian WB, Wu LL, Huang ZN, Hu X, et al. Serum Klotho, vitamin D, and homocysteine in combination predict the outcomes of Chinese patients with multiple system atrophy. CNS Neurosci Ther 2017;23:657–666.ArticlePubMedPMC
- 97. Kasai T, Tokuda T, Ohmichi T, Ishii R, Tatebe H, Nakagawa M, et al. Serum levels of coenzyme Q10 in patients with multiple system atrophy. PLoS One 2016;11:e0147574.ArticlePubMedPMC
- 98. Herbert MK, Eeftens JM, Aerts MB, Esselink RA, Bloem BR, Kuiperij HB, et al. CSF levels of DJ-1 and tau distinguish MSA patients from PD patients and controls. Parkinsonism Relat Disord 2014;20:112–115.ArticlePubMed
- 99. Hansson O, Janelidze S, Hall S, Magdalinou N, Lees AJ, Andreasson U, et al. Blood-based NfL: a biomarker for differential diagnosis of parkinsonian disorder. Neurology 2017;88:930–937.ArticlePubMedPMC
- 100. Low PA, Robertson D, Gilman S, Kaufmann H, Singer W, Biaggioni I, et al. Efficacy and safety of rifampicin for multiple system atrophy: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2014;13:268–275.ArticlePubMedPMC
- 101. Poewe W, Seppi K, Fitzer-Attas CJ, Wenning GK, Gilman S, Low PA, et al. Efficacy of rasagiline in patients with the parkinsonian variant of multiple system atrophy: a randomised, placebo-controlled trial. Lancet Neurol 2015;14:145–152.ArticlePubMed
- 102. Rascol O. Preliminary results of the French MSA-fluoxetine study: P1469. Eur J Neurol 2012;19 Suppl 1:262.
- 103. Saccà F, Marsili A, Quarantelli M, Brescia Morra V, Brunetti A, Carbone R, et al. A randomized clinical trial of lithium in multiple system atrophy. J Neurol 2013;260:458–461.ArticlePubMedPDF
- 104. Ozawa T, Tada M, Kakita A, Onodera O, Tada M, Ishihara T, et al. The phenotype spectrum of Japanese multiple system atrophy. J Neurol Neurosurg Psychiatry 2010;81:1253–1255.ArticlePubMed
- 105. Ozawa T. The COQ2 mutations in Japanese multiple system atrophy: impact on the pathogenesis and phenotypic variation. Mov Disord 2014;29:184.ArticlePubMed
- 106. Kim HJ, Stamelou M, Jeon B. Multiple system atrophymimicking conditions: diagnostic challenges. Parkinsonism Relat Disord 2016;22 Suppl 1:S12–S15.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
- Cranial Nerve Thinning Distinguishes RFC1‐Related Disorder from Other Late‐Onset Ataxias
Camila C. Lobo, Guilherme S.O. Wertheimer, Gabriel S. Schmitt, Paula C.A.A.P. Matos, Thiago J.R. Rezende, Joyce M. Silva, Fabrício C. Borba, Fabrício D. Lima, Alberto R.M. Martinez, Orlando G.P. Barsottini, José Luiz Pedroso, Wilson Marques, Marcondes C.
Movement Disorders Clinical Practice.2024; 11(1): 45. CrossRef - The potential of phosphorylated α‐synuclein as a biomarker for the diagnosis and monitoring of multiple system atrophy
Toufik Abdul‐Rahman, Ranferi Eduardo Herrera‐Calderón, Arjun Ahluwalia, Andrew Awuah Wireko, Tomas Ferreira, Joecelyn Kirani Tan, Maximillian Wolfson, Shankhaneel Ghosh, Viktoriia Horbas, Vandana Garg, Asma Perveen, Marios Papadakis, Ghulam Md Ashraf, Ath
CNS Neuroscience & Therapeutics.2024;[Epub] CrossRef - The effect of continuous positive airway pressure (CPAP) on the quality of life in patients with multiple system atrophy
Hee Jin Chang, Han-Joon Kim, Kyung Ah Woo, Jung Hwan Shin, Ki-Young Jung
Sleep and Breathing.2023; 27(4): 1481. CrossRef - Psychosis treatment in a patient with Parkinsonian type multiple system atrophy using modified electroconvulsive therapy: a case report
Takumi Yawata, Shunsuke Takagi, Takehiro Tamura, Genichi Sugihara, Hidehiko Takahashi
Psychogeriatrics.2023; 23(2): 364. CrossRef - Comparison of the second consensus statement with the movement disorder society criteria for multiple system atrophy: A single-center analysis
Yunchuang Sun, Wei Sun, Luhua Wei, Fan Li, Yanyan Jiang, Fei Zhai, Mingyue Luan, Jing Chen, Zhaoxia Wang
Parkinsonism & Related Disorders.2023; 106: 105242. CrossRef - Multiple System Atrophy: Advances in Diagnosis and Therapy
Hirohisa Watanabe, Sayuri Shima, Yasuaki Mizutani, Akihiro Ueda, Mizuki Ito
Journal of Movement Disorders.2023; 16(1): 13. CrossRef - Multiple system atrophy-cerebellar: A case report and literature review
Thi Thuong Doan, Thuy Dung Pham, Duy Duan Nguyen, Dac Hong An Ngo, Trong Binh Le, Thanh Thao Nguyen
Radiology Case Reports.2023; 18(3): 1121. CrossRef - Hjerne med kors
Linh Tran, Tuba Ahmad, Anniken Haslund, Phuoc Ngoc Thi Nguyen
Tidsskrift for Den norske legeforening.2023;[Epub] CrossRef - α-Synuclein Strains and Their Relevance to Parkinson’s Disease, Multiple System Atrophy, and Dementia with Lewy Bodies
Noah J. Graves, Yann Gambin, Emma Sierecki
International Journal of Molecular Sciences.2023; 24(15): 12134. CrossRef - A Rare Case of Cerebellar Ataxia
Aanchal Arora, Nidhi Hooda, JasneetSingh Channa, Motilal Negi
Neurology India.2023; 71(5): 1031. CrossRef - Neurodegenerative disorders affecting the autonomic nervous system: Pure autonomic failure and multiple system atrophy
Yoshitaka Yamanaka, Nobuyuki Araki, Satoshi Kuwabara
Neurology and Clinical Neuroscience.2022; 10(3): 115. CrossRef - Glia Imaging Differentiates Multiple System Atrophy from Parkinson's Disease: A Positron Emission Tomography Study with [11C]PBR28 and Machine Learning Analysis
Aurelija Jucaite, Zsolt Cselényi, William C. Kreisl, Eugenii A. Rabiner, Andrea Varrone, Richard E. Carson, Juha O. Rinne, Alicia Savage, Magnus Schou, Peter Johnström, Per Svenningsson, Olivier Rascol, Wassilios G. Meissner, Paolo Barone, Klaus Seppi, Ho
Movement Disorders.2022; 37(1): 119. CrossRef - Data-driven subtype classification of patients with early-stage multiple system atrophy
Hui-Jun Yang, Han-Joon Kim, Yu Jin Jung, Dallah Yoo, Ji-Hyun Choi, Jin Hee Im, Beomseok Jeon
Parkinsonism & Related Disorders.2022; 95: 92. CrossRef - Clinicopathological correlates of pyramidal signs in multiple system atrophy
Chi‐Ying R. Lin, Anisha Viswanathan, Tiffany X. Chen, Hiroshi Mitsumoto, Jean P. Vonsattel, Phyllis L. Faust, Sheng‐Han Kuo
Annals of Clinical and Translational Neurology.2022; 9(7): 988. CrossRef - The “Black Straight-Line Sign” in the Putamen in Diffusion-Weighted Imaging: A Potential Diagnostic MRI Marker for Multiple System Atrophy
Yiming Zheng, Xiwen Wang, Huajian Zhao, Yanyan Jiang, Ying Zhu, Jing Chen, Wei Sun, Zhaoxia Wang, Yunchuang Sun
Frontiers in Neurology.2022;[Epub] CrossRef - Clinical Aspects of the Differential Diagnosis of Parkinson’s Disease and Parkinsonism
Hae-Won Shin, Sang-Wook Hong, Young Chul Youn
Journal of Clinical Neurology.2022; 18(3): 259. CrossRef - Clinical Features and Neuroimaging Findings of Neuropil Antibody–Positive Idiopathic Sporadic Ataxia of Unknown Etiology
Akira Takekoshi, Akio Kimura, Nobuaki Yoshikura, Isamu Yamakawa, Makoto Urushitani, Katsuya Nakamura, Kunihiro Yoshida, Takayoshi Shimohata
The Cerebellum.2022; 22(5): 915. CrossRef - Determinants of cognitive impairment in multiple system atrophy: Clinical and genetic study
Amina Nasri, Alya Gharbi, Ikram Sghaier, Saloua Mrabet, Amira Souissi, Amina Gargouri, Mouna Ben Djebara, Imen Kacem, Riadh Gouider, Tuhin Virmani
PLOS ONE.2022; 17(12): e0277798. CrossRef - Management of balance problems in an elderly with multiple system atrophy with predominant cerebellar ataxia (MSA-C) and sick sinus syndrome
Amber Eker, Pembe Hare Yıgıtoglu, Hamza Duygu, Ersin Tan
Journal of Gerontology and Geriatrics.2021; 69(3): 208. CrossRef - Immunotherapies in Huntington's disease and α-Synucleinopathies
Oluwaseun Fatoba, Yosuke Ohtake, Takahide Itokazu, Toshihide Yamashita
Frontiers in Immunology.2020;[Epub] CrossRef - The Dysfunctional Autonomic Function and “Dysfunctional” Fatigue in Drug Naïve Parkinson’s Disease
Jong Hyeon Ahn, Minkyeong Kim, Jun Kyu Mun, Yoonsu Cho, Ji Sun Kim, Jinyoung Youn, Joong-Seok Kim, Jin Whan Cho
Journal of Parkinson's Disease.2020; 10(2): 605. CrossRef - Early autonomic and cognitive dysfunction in PD, DLB and MSA: blurring the boundaries between α-synucleinopathies
Giovanni Palermo, Eleonora Del Prete, Ubaldo Bonuccelli, Roberto Ceravolo
Journal of Neurology.2020; 267(12): 3444. CrossRef - Changes of Amide Proton Transfer Imaging in Multiple System Atrophy Parkinsonism Type
Shuhua Li, Piu Chan, Chunmei Li, Haibo Chen, Min Chen, Wen Su, Kai Li, Na Lu, Lu Yu, Defa Chu, Pu-Yeh Wu
Frontiers in Aging Neuroscience.2020;[Epub] CrossRef - Frontline of Non-genetic Cerebellar Ataxia
Nobuaki Yoshikura, Akio Kimura, Takayoshi Shimohata
Nihon Naika Gakkai Zasshi.2020; 109(6): 1138. CrossRef - Purva Rupeeyam of bhela indriya sthana-an explorative study
Kshama Gupta, Prasad Mamidi
International Journal of Complementary & Alternative Medicine.2020; 13(6): 228. CrossRef - A case of multiple system atrophy
Jing Guo, Fuying Liu, Tingting Liu, Xin Zhang, Yong Luo
Journal of International Medical Research.2019; 47(11): 5839. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite