 E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 12(2); 2019 > Article
-
Review Article
Altered Gut Microbiome and Intestinal Pathology in Parkinson’s Disease -
Han-Lin Chiang1
 , Chin-Hsien Lin2
, Chin-Hsien Lin2 -
Journal of Movement Disorders 2019;12(2):67-83.
DOI: https://doi.org/10.14802/jmd.18067
Published online: May 30, 2019
1Department of Neurology, Neurological Institute, Taipei Veterans General Hospital, Taipei, Taiwan
2Department of Neurology, National Taiwan University Hospital, College of Medicine, National Taiwan University, Taipei, Taiwan
- Corresponding author: Chin-Hsien Lin, MD, PhD Department of Neurology, National Taiwan University Hospital, No.7 Chuang-Shan South Road, Taipei 100, Taiwan / Tel: +886-2-23123456 / Fax: +886-2-23418395 / E-mail: chlin@ntu.edu.tw
Copyright © 2019 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Parkinson’s disease (PD) is a common neurodegenerative disorder arising from an interplay between genetic and environmental risk factors. Studies have suggested that the pathological hallmarks of intraneuronal α-synuclein aggregations may start from the olfactory bulb and the enteric nervous system of the gut and later propagate to the brain via the olfactory tract and the vagus nerve. This hypothesis correlates well with clinical symptoms, such as constipation, that may develop up to 20 years before the onset of PD motor symptoms. Recent interest in the gut–brain axis has led to vigorous research into the gastrointestinal pathology and gut microbiota changes in patients with PD. In this review, we provide current clinical and pathological evidence of gut involvement in PD by summarizing the changes in gut microbiota composition and gut inflammation associated with its pathogenesis.
- Gastrointestinal (GI) impairments are commonly observed at all stages of PD, with almost 30% of patients reporting GI symptoms, including drooling, dysphagia, gastroparesis, and constipation [18]. The prevalence of drooling in PD ranges from 10% to 84% [18-22]. The presentation is related to swallowing dysfunction in the oropharyngeal phase [19] and an increased rate of parotid gland secretion [20] that worsens with a flexed posture, unintended mouth opening, and divided attention [21]. The prevalence of dysphagia ranges from 9% to 82% but has been up to 97% in objective studies [22]. Dysphagia usually develops in patients with advanced PD who have severe bradykinesia and rigidity, which is thought to contribute to oropharyngeal dysphagia [23]. The prevalence of gastroparesis ranges from 70% to 100%, with an average half-emptying time of 46 to 149 minutes in patients with mild PD and 55 to 221 minutes in moderate/severe PD, compared to 43 to 107 minutes in healthy controls [24]. Although the exact pathophysiology is unclear, gastroparesis is a major player in the development of motor fluctuations in PD [25].
- The most common GI symptom in patients with PD is constipation, which is also among the most prevalent prodromal symptoms of PD [26]. The prevalence of constipation in PD ranges from 8% to 70% and increases with disease progression [27]. Evidence suggests that this prevalence is up to 6‑fold greater among patients with PD than among age-matched and sex-matched controls [28]. An increasing number of studies have evaluated the temporal relationship between bowel movement frequency and PD risk. The Honolulu-Asia Aging cohort study in males, which had a 24-year follow-up period, showed that infrequent bowel movements, as assessed by a self-reported bowel habit questionnaire, were associated with an increased risk of PD and reduced neuronal density in the substantia nigra [29]. Our study using a population-based national database also showed that the severity of constipation, quantitatively defined by the amount of laxative use, was dose-dependently associated with future PD risk, independent of age, sex, underlying comorbidities, or medication use [30]. A case–control study reported a greater PD risk among individuals with a history of constipation, as early as 20 years before the onset of motor symptoms, as assessed by a review of medical records [31]. PD-related constipation may be traced to LB deposition in the ENS or the dorsal motor nucleus of the vagus nerve, which are among the earliest affected regions in PD [32]. Despite vigorous research, the precise etiology of constipation and whether constipation in PD is caused by gut or brain pathology remain elusive [33].
GASTROINTESTINAL SYMPTOMS IN PATIENTS WITH PD
- The GI tract consists of four layers: the mucosa, submucosa, muscular layer, and serosa (ordered from innermost to outermost). It contains its own intrinsic nervous system, the ENS, but is also modulated extrinsically by the autonomic nervous system, which is involved in PD [34]. The ENS consists of submucosal plexuses and myenteric (Auerbach) plexuses (Figure 1). The submucosal plexuses have three layers: the inner (Meissner plexus), intermediate, and outer (Schabadasch or Henle) plexuses, which are mainly responsible for secretion and blood flow in the GI tract. Myenteric plexuses mostly control intestinal peristalsis [35]. Virtually every neurotransmitter produced by neurons in the central nervous system (CNS) have also been identified within the ENS, including acetylcholine, nitric oxide (NO), vasoactive intestinal peptide (VIP), and catecholamines [36]. Autonomic parasympathetic input mediates the central modulation of ENS function primarily from the dorsal motor nucleus of the vagus nerve and the sympathetic input from the para- and prevertebral ganglia.
- The accumulation of α-synuclein is not limited to central dopaminergic neurons, and a growing body of literature has shown that α-synuclein aggregation can be detected particularly in the ENS at postmortem and in safely accessible in vivo biopsies of the GI tract. Some studies have even demonstrated detectable α-synuclein pathology in the GI tract in the prodromal phase of PD. Below, we summarize the recent GI pathology findings in patients with PD according to the findings from postmortem pathology studies, colon biopsy (only covering the mucosal and submucosal layers) and surgical colectomy findings (covering all three layers of colon) of PD patients.
- Evidence from postmortem autopsy studies
- In 1984, Qualman and colleagues [37] first reported the possible presence of LBs in the GI tract, based on the autopsies of 8 patients with achalasia, 22 with PD, and 50 controls. Using hematoxylin-eosin (H&E) staining, they examined multiple levels of the entire GI tract (esophagus, stomach, jejunum, ileum colon, and rectum). LB-like intraneuronal inclusions were found in the myenteric plexus of the esophagus in 2 of 8 patients with achalasia and 1 of 3 PD patients with dysphagia, but none were found in the controls. This intraneuronal inclusion pathology was most prominent in the distal esophagus in the achalasia patient who had the shortest duration of the disease and retained degenerated ganglion cells in the esophagus. The inclusions were also found in the myenteric plexus but not the submucosal plexus of the colon in one PD patient who had dysphagia. Subsequent autopsy reports by Wakabayashi and colleagues [38,39] using H&E staining and electron microscopy described LB-like inclusions in both the myenteric plexus and submucosal plexus and throughout the GI tract in PD patients, more numerously in the distal esophagus. Intriguingly, small LBs were also observed in the myenteric plexus of the esophagus or small intestine in 8 of 24 age-matched controls who did not have PD [39]. Further immunohistochemistry staining of different types of neurons of 3 autopsied PD patients showed that the LBs resided in the VIP neurons instead of tyrosine hydroxylase neurons in the paravertebral and celiac sympathetic ganglia and the ENS of the GI tract [38].
- In 2006, Braak and colleagues [40] investigated the gastric pathology of 5 patients with pathologically confirmed PD using immunohistochemistry with an antibody against α-synuclein (anti-Syn-1, clone 42). α-synuclein pathology, in the form of LBs and immunoreactive fibers, was identified in the myenteric plexus, submucosal plexus, and peripheral nerves in the adventitia of the distal esophagus and stomach in all PD patients (5/5) but in no controls (0/5) (Table 1). One of the five PD patients had α-synuclein pathology in the ENS and dorsal motor nucleus of the vagal nerve without α-synuclein deposits in the substantia nigra. These findings, in combination with the same group’s discovery of an α-synuclein staging pattern in the CNS [32], led to Braak and colleagues’ pathology hypothesis of PD [3,4] and opened the door to subsequent vigorous research on α-synuclein in the gut.
- The autopsy reports of immunohistochemistry staining targeting α-synuclein in the GI tracts are summarized in Table 1. Most studies used antibodies against α-synuclein, although with different clones. The frequency of α-synuclein detection in PD patients was higher than in controls in most studies (Table 1) [40-45]. One group using an antibody targeting phosphorylated Ser 129 α-synuclein obtained positive results in 11 of 17 patients with PD and in 0 of 23 controls [46]. In addition to patients with PD, α-synuclein pathology was observed in patients with diffuse LB dementia (DLB), comprising 5 of 9 patients with DLB in one study and 5 of 5 in another; on the other hand, it was rarely found in patients with Alzheimer’s disease [44,45] and not found in 2 patients with multiple system atrophy in a study that examined only the esophagus [43].
- These autopsy reports using immunohistochemistry to target intraneuronal α-synuclein were in agreement with a rostro–caudal gradient pattern of α-synuclein distribution in the GI tract, with more abundant α-synuclein pathology in the esophagus and stomach compared to the colon [41,44,46]. In addition, α-synuclein pathology was found in both the myenteric and submucosal plexuses but more frequently in the former. LBs, if present, were mostly found in the myenteric plexus. Notably, Annerino and colleagues [41] reported that in contrast to earlier findings of Wakabayashi and colleagues, α-synuclein pathology did not colocalize with NO-positive or VIP-positive neurons and did so with less than 3% of the tyrosine hydroxylase–positive neurons. PD motor symptom severity also did not correlate with the density of enteric neurons or α-synuclein pathology in that study [41].
- Evidence from colon biopsy studies
- Following the initial autopsy reports of α-synuclein in the GI tract [40], many studies addressed the feasibility of detecting α-synuclein as a peripheral biomarker using colonic biopsy specimens of living patients. The results of the biopsy reports are summarized in Table 2. Overall, they were inconsistent. Because of safety concerns, the specimens from a biopsy procedure contain only the mucosa and, at most, the submucosa. Based on immunohistochemistry, the frequency of α-synuclein or phospho-α-synuclein in the stomach mucosa ranged from 31.6% to 100% in PD patients and 4.3% to 41.7% in controls [47-49]; the frequency of α-synuclein or phospho-α-synuclein in the mucosal layer of the colon was 0% to 100% in PD patients and in controls [47,48,50-53]; and the frequency of α-synuclein or phospho-α-synuclein in the submucosal layer of the colon was 5% to 100% in PD patients and 0% to 100% in controls [52-59]. Some groups examined α-synuclein or phospho-α-synuclein in various regions of the mucosa or mucosal layers of the GI tract and found frequencies from 9% to 100% in PD patients and 0% to 36% in controls [60-64]. Although the findings of many studies still supported a higher frequency of α-synuclein or phospho-α-synuclein in PD patients than in controls [49,53-59,61,62], others suggested similar frequencies between groups [47,48,50-52,60,63-65].
- Despite the higher frequency of α-synuclein or phospho-α-synuclein deposits in colonic biopsy specimens in PD patients versus controls in some studies, others offered conflicting results (Table 2). Some explanations for the discrepancies include the variable individual GI conditions of participants, different PD subtypes and disease durations, variable location and depth of the biopsies, the number of biopsies and slides examined, specimen size, different definitions for α-synuclein positivity, different neuronal markers for nerves and neurons, different antibodies, and most important, different specimen preparation (e.g., whole-mount with microdissection vs. formalin-fixed, paraffin-embedded) and immunohistochemistry methods.
- Although some studies uncovered a rostro–caudal gradient of α-synuclein deposits in colonic biopsies [47,57,62], other studies did not [52,61]. α-synuclein pathology can also be found in the preclinical stage of PD patients [58,61] and in patients with idiopathic REM sleep behavior disorder [52]. The proportion of α-synuclein positivity did not differ between patients with preclinical PD and those with motor symptoms of PD in one study (10.6% were in the preclinical stage, defined as up to 8 years prior to clinical diagnosis; 10.7% had PD motor symptoms) [61]. The difference between preclinical PD and controls was detected only with an antibody against phospho-α-synuclein in another study (phospho-α-synuclein positivity in PD vs. preclinical PD vs. controls: 48%, 45%, and 26%, respectively; total α-synuclein positivity in PD vs. preclinical PD vs. controls: 61%, 52%, and 47%, respectively) [65]. Additionally, biopsy positivity did not differ between patients in the early and late stages of PD [51].
- Few studies have evaluated the correlation between motor symptom severity and α-synuclein pathology of the GI tract. Lebouvier and colleagues [56] reported a positive correlation of axial symptom subscores of the unified PD rating scale (UPDRS), levodopa responsiveness, and chronic constipation severity with LB pathology in the submucosa of the GI tract. Hilton and colleagues [61] found that all PD patients with positive findings of α-synuclein pathology in colonic biopsies had early autonomic symptoms, although other findings conflicted with these results [49,47,63].
- Evidence from surgical specimens of colectomy
- Surgical specimens are superior to biopsy specimens because they allow for better specimen quality with adequate tissue and full thickness of the intestinal wall. However, the disadvantage of studies using surgical specimens is that only diseased specimens can be obtained, which can affect the results. The results of reports based on surgical specimens are summarized in Table 3. Shin and colleagues [64] compared phospho-α-synuclein positivity (EP1536Y) in colonic biopsy specimens with surgical specimens from patients with PD. Of interest, although the stomach surgical specimens had a higher positive rate for phospho-α-synuclein immunoreactivity in PD participants (58%) than in controls (8.3%), the colorectal surgical specimens did not differ (PD patients vs. controls: 23.8% vs. 23.8%). Furthermore, the positivity of colorectal mucosal biopsy specimens did not differ between PD patients (9.1%) and controls (18.2%) [64]. In comparison studies between PD patients and controls, the overall frequency of α-synuclein pathology in PD patients detected using surgical specimens has been 36% to 100%, higher than in the controls (0% to 22%) [64,66-68].
- Yan and colleagues [68] studied surgical specimens, including colon and stomach, using an α-synuclein mouse monoclonal antibody and found that α-synuclein pathology is most frequent in the body of the stomach in PD patients but not in controls. Using different primary antibodies, Aldecoa and colleagues [66] detected LB-like intraneuronal aggregations in the body of the stomach in 4 out of 6 PD patients but in only 1 out of 12 controls. Additionally, the pattern of punctate cytoplasmic staining of ganglion cells was observed only in staining with anti-phosphorylated Ser129 α-synuclein antibody [66]. However, some studies have produced conflicting results, identifying α-synuclein pathology in the submucosal and muscular layers of the colon or appendiceal mucosal nerve plexus in neurologically healthy controls who underwent partial colectomy or sigmoid resection [69,70].
- These conflicting results have led to several investigations into the sensitivity and specificity of different conventional immunohistochemistry methods or to attempts with other methods to produce more consistent results. Corbillé and colleagues [71] distributed a common set of colon biopsy slides from 9 PD patients and 3 controls to four different laboratories for immunohistochemistry staining using different α-synuclein or phospho-α-synuclein antibodies and epitope exposure methods. Their results showed that no particular single staining method or staining pattern had a sensitivity and specificity of more than 80%. The same group further examined full-thickness sigmoid colon specimens from 5 pathology-confirmed PD patients and 5 controls using different immunohistochemistry methods and antibodies [anti-phospho-Ser129 α-synuclein, including EP1536Y (ab51253) and MJF-R13 (8-8) (ab168381), anti-phospho-Ala81 α-synuclein, polyclonal p-synuclein LB509], and epitope exposure methods [72]. They obtained excellent accuracy with 100% specificity and sensitivity most frequently in the submucosa instead of the muscularis layer with the combination of a specific staining pattern (“fibers and puncta” and “fibers only” representing neuronal distribution), a particular method with primary antibody targeting anti-phospho-Ser129 α-synuclein {[MFJ-R13 (8-8)] ab168381 with a concentration of 1:5,000, and an epitope exposure method using proteinase K with a concentration of 1:100 in 37°C for 20 minutes}, and raters with good interrater agreement [72]. Therefore, an immunohistochemistry study for assessing α-synuclein pathology in the colon is reliable only if adequate submucosa tissue is obtained, a larger amount of α- synuclein pathology is present, a specific staining pattern associated with neuronal morphology is recognized, a suitable immunohistochemistry method is used, and raters are well trained [72].
- Punsoni and colleagues [73] performed quantitative polymerase chain reaction (qPCR) in addition to conventional immunohistochemistry methods in 4 groups of participants, including living PD patients, PD autopsy cases, living pediatric controls, and living adult controls. The specimens included different regions of the GI tract and of different thicknesses, including submucosa and myenteric plexuses. The expression of α-synuclein in neurons was confirmed by colocalization with neurofilament staining. Immunohistochemistry analysis showed that although α-synuclein staining was diffusely present in all PD patients and controls, LBs were observed in the myenteric plexus of 25% of PD patients but not in controls. Moreover, the mean α-synuclein expression examined by qPCR was highest in PD patients and lowest in controls [73]. Visanji and colleagues [51] used proteinase K to enhance antigen retrieval in immunohistochemistry analysis in paraffin-embedded tissues and found a lower positive rate of either total or phospho-α-synuclein immunoreactivity in PD patients than in controls. Barrenschee and colleagues [60] used morphometric analysis followed by dual-label immunohistochemistry and found that the total numbers and areas of phospho-α-synuclein aggregates per neuron were increased in PD patients compared with controls.
- Summary of gastrointestinal pathology findings in patients with PD
- In summary, no consensus technique has yet been established for detecting α-synuclein pathology in gut tissues that can reliably differentiate PD patients from controls. To allow clinicians to use α-synuclein pathology staining in the GI tract as a biomarker for PD pathogenesis, the development or testing of other techniques is warranted. This process should include both quantitative and qualitative methods in addition to careful stratification of PD patients into different subtypes, focusing on those who are most likely to be in line with Braak and colleagues’ hypothesis.
GASTROINTESTINAL PATHOLOGY FINDINGS IN PATIENTS WITH PD
- Neuronal loss in the ENS in PD
- In the first autopsy report of LBs in the ENS using H&E staining, degeneration of enteric ganglion cells was found in the myenteric plexus in the esophagus of one patient with PD who had dysphagia [37]. A subsequent autopsy report of one patient with PD and acquired megacolon found well-preserved ganglia and ganglion cells in both the submucosal and myenteric plexuses of the colon using H&E staining [74]. Neuron counting via immunohistochemistry or coimmunofluorescence staining also showed variable results in both the submucosa and the muscular layer. Lebouvier and colleagues [55,75] examined colonic biopsy specimens with the mouse pan-neuronal marker anti-Hu C/D and found no difference between PD patients and controls in the neuron number per ganglion or proportion of dopaminergic neurons in the submucosal layer. However, their subsequent study showed a decreased number of neurofilament heavy chainimmunoreactive neurons per ganglion in patients with positive phosphorylated α-synuclein aggregations in the submucosa. Furthermore, the LB pathology burden was negatively correlated with neuronal count and levodopa responsiveness but positively correlated with axial symptoms and constipation severity [56]. On the other hand, a recent study examining rectal biopsy specimens using the pan-neuronal marker PGP9.5 showed that patients and controls had similar mean neuronal area, ganglionic area, and neuron number per ganglion in the submucosa of rectum [60]. The divergent findings in these studies are probably the result of the randomly biopsied regions and different methods used. Therefore, although most patients with PD have constipation, it may not be directly related to neuronal loss in the ENS; instead, the extrinsic autonomic system probably plays a role [34].
- Evidence of enteric glial cells involved in PD
- Enteric glial cells (EGCs) are another population of cells in the ENS known for a role in neuroprotection, gut inflammation, intestinal epithelial barrier function, and synaptic neurotransmission regulation [76]. As there was no overt neuronal loss identified in the ENS, another possible cause of GI dysfunction may be traced to the EGCs. Despite a large variation among patients, two studies suggested that reactive gliocytosis (severe glia reactions) might be present in the bowel given the general increase in the expression of the glial markers, glial fibrillary acidic protein and Sox-10 [77,78]. The underlying mechanism of these phenomena remains elusive, and further studies are warranted to elucidate the involvement of EGCs in the PD process.
- Evidence of leaky gut and bowel inflammation in PD
- Recent animal studies have shown that bowel inflammation can exacerbate neuroinflammation, disrupt the blood–brain barrier, and promote dopaminergic neuronal loss in the substantia nigra of an inflammation rat model of PD. In this model, substantia nigra inflammation and selective dopaminergic neuronal loss were induced by LPS injection [79]. In agreement, several studies have shown clinical evidence of bowel inflammation in PD patients. Devos and colleagues [78] used real-time PCR to analyze the mRNA expression of pro-inflammatory cytokines and glial markers in the ascending colon biopsies of PD patients and controls. They found significantly increased expression of proinflammatory cytokines [tumor necrosis factor alpha (TNF-α), interferon gamma (IFN-γ), interleukin (IL)-6, and IL-1β] in PD patients. This increase correlated with increased expression of glial markers (glial fibrillary acidic protein and Sox-10), indicating that pro-inflammatory processes in the bowel are enhanced in PD patients. Of note, the authors found that expression levels of cytokines and glial markers did not correlate with immunostaining levels of phospho-α-synuclein and axial symptom subscores on the UPDRS part III. In addition, these expression levels correlated negatively with PD duration. This observation suggests that bowel inflammation may play a role in PD pathogenesis [78].
- Consistent with this possibility, stool immune profiles show elevations in proteins related to angiogenesis and in chemokines and cytokines such as IL-1α, IL-1β, and IL-8 in PD patients compared to controls [80]. Another study also reported increases in the fecal intestinal permeability marker fecal calprotectin in patients with PD compared to age-matched controls; however, the level of fecal calprotectin did not correlate with any clinical parameters, including disease duration, in patients with PD [81].
- Recent evidence suggests that gut inflammation in PD may be traced to increased intestinal permeability, also known as the leaky gut. This status correlates with intestinal α-synuclein accumulation in patients with PD [82]. Increased intestinal permeability and translocation of bacteria and inflammatory bacterial products (e.g., LPS) might lead to inflammation in the GI tract and thereby initiate α-synuclein accumulation in the ENS [82]. Forsyth and colleagues [82] also observed increased urine sucralose excretion in patients with PD compared to controls, suggesting increased colon permeability. Furthermore, increased colon permeability correlates with increased α-synuclein accumulation pathology and E. coli immunohistochemistry staining of distal sigmoid biopsy in PD patients [83]. This observation supports the hypothesis that increased permeability-related bowel inflammation with an increased chance of gut bacteria translocation may be involved in PD pathogenesis.
- Clairembault and colleagues [54] further examined morphological changes and expression in the intestinal epithelial barrier (including two tight junction proteins, ZO-1 and occludin) in colonic biopsy tissues of PD patients and controls. They found that a larger proportion of PD patients (14 of 31 patients) had disrupted and irregularly distributed tight junction proteins and lower expression levels of occludin compared with controls (1 of 8 controls) [54]. These observations provide some evidence of increased gut permeability and mild bowel inflammatory changes observed in patients with PD.
- The link between bowel inflammation and PD also comes from recent findings of common genetic risk variants between inflammatory bowel disease (IBD) and PD [84]. Among these candidates are two genes, NOD2 and LRRK2, the latter of which is the most common causative genetic variant linked to PD. They are of special interest because of their involvement in the innate immune system, which regulates the immune response [85]. Genetic findings have led to several epidemiological studies examining the relationship of PD risk in patients with IBD. Four large population studies from Taiwan (person-years: 299,900) [86], Denmark (person-years: > 8,000,000) [87], the United States (personyears: 3,008,825) [88], and Sweden (person-years: 249,784) [89] have demonstrated increased PD risk in patients with IBD. PD incidence thereby increased by 35, 22, 28, and 30% in patients with IBD in the Taiwanese, Danish, American, and Swedish populations, respectively [86-89]. The risk was higher in patients with Crohn’s disease (CD) than in those with ulcerative colitis (UC) in the Taiwanese population but higher in those with UC than in those with CD in the Swedish and Danish studies [87,89]. In the US study, the PD risk increase was similar for CD and UC [86-89].
- Consistently, two US studies also showed that treatment of IBD, one with anti-TNF therapy [88] and the other with unspecified immunosuppressants [90], reduced PD risk. In the Swedish study, patients with IBD who did not receive treatment were 60% more likely to develop PD than their matched controls, whereas treated IBD patients had a nonsignificantly decreased risk of developing PD compared to nontreated patients [89]. However, a retrospective study enrolling 876 patients with PD identified only 2 patients with CD prior to the PD diagnosis, an incidence similar to that in the general population [91]. Another large population-based, case–control study enrolling patients aged 65 years or older with newly diagnosed PD (n = 89,790) and controls (n = 118,095) identified an inverse correlation between PD and IBD, suggesting that patients with IBD are less likely to develop PD [90].
- Despite the inconsistent results, these recent pathological, genetic, and epidemiological studies have suggested a relationship between bowel inflammation and PD. The exact mechanism linking bowel inflammation and neurodegeneration in the disease process of PD is still elusive and requires further studies in the future, which would shed light on potential treatment strategies for halting the neurodegenerative process of PD.
MECHANISM OF GI IMPAIRMENT AND GUT PATHOLOGY IN PD
- Gastric Helicobacter pylori and small intestinal microbial overgrowth
- Gastric Helicobacter pylori infection is a common chronic infection that has been implicated in PD [92-101]. Most epidemiological studies using different detection methods (ELISA, urea breath test, RT-PCR, histological examination) have shown an increased risk of H. pylori infection in PD, ranging from 1.3- to 2-fold (prevalence: 22% to 70%) [92-101], which was also shown to be significant in a recently published meta-analysis [102]. Meta-analyses have also identified a significantly worse mean UPDRS score in PD patients with H. pylori infection (either UPDRS or total UPDRS III in “on” or “off” state) [97,98,103-107] and improvement in UPDRS part III scores after H. pylori eradication [102-105,108,109]. The exact mechanisms of involvement of chronic H. pylori infection in PD pathogenesis are unclear but possibly trace to multifactorial inputs, including the H. pylori toxin, neuroinflammation, and alterations in the gut microbiome [110].
- Small intestinal bacterial overgrowth (SIBO) is also more prevalent in patients with PD than in healthy controls in crosssectional studies, with a prevalence of 25% to 54% [97,111-114]. Most studies used lactulose or glucose breath tests to detect SIBO, with sensitivities of 31–68% and 29–93%, respectively, and specificities of 44–100% and 30–86%, respectively [115]. Bloating and flatulence [112,113] have been reported to be associated with SIBO in PD in some studies, whereas constipation and tenesmus were reported in one study [114]. The relationship between PD symptoms and SIBO is heterogeneous among reports. Disease duration, Hoehn and Yahr stage, UPDRS, motor scores, motor fluctuations, or nonmotor symptom severity have all been mentioned in association with SIBO [97,112-114]. Treatment of SIBO is reported to have improved motor fluctuation symptoms in PD patients, but the recurrence rate at 6 months was as high as 43% [97].
- Altered gut microbiota in PD
- To date, 13 studies (including one published abstract) have compared the difference in gut microbiota composition between PD patients and controls (Table 4 and 5) [116-128]. While α-diversity (species diversity within a single subject) was similar in most studies [116,120,122,123,126,127], two showed increased [121,125] and two showed decreased α-diversity [124]. All studies revealed differences in microbiota composition between PD patients and controls (β-diversity), but the results were heterogeneous (Table 4). In brief, when compared with controls, the family Verrucomicrobiaceae (phylum Verrucomicrobia) and genera Akkermansia (phylum Verrucomicrobia; family Verrucomicrobiaceae) and Lactobacillus (phylum Firmicutes; family Lactobacillaceae) were increased in PD patients in several studies [116,118,119,126]. On the other hand, the families Prevotellaceae (phylum Bacteroidetes), Lachnospiraceae (phylum Firmicutes), and Pasteurellaceae (phylum proteobacteria) and genera Blautia (phylum Firmicutes; family Lachnospiraceae), Roseburia (phylum Firmicutes; family Lachnospiraceae), Prevotella (phylum Bacteroidetes; family Prevotellaceae), and Faecalibacterium (phylum Firmicutes; family Clostridiaceae) were decreased in PD patients in three or more studies [116,126,128]. Prevotella is abundant in humans that consume plant-based, fiber-rich diets, which are substrates for bacteria to produce short chain fatty acids (SCFAs) [129]. Prevotella and Akkermansia are both mucin-degraders that promote and serve as indicators of gut integrity [130]; however, they both have also been linked to gut or systemic inflammatory conditions [129,131]. On the other hand, Faecalibacterium prausnitzii has anti-inflammatory properties other than an effect of butyrate production [132-134]. Of interest, similar to PD patients, Faecalibacterium prausnitzii and Lachnospiraceae are depleted in patients with IBDs. In contrast to PD patients, however, Akkermansia is decreased in patients with IBDs [135].
- Some groups have tried to delineate the relationship between fecal microbiota and clinical features but with heterogeneous results. Two studies identified a correlation of PD duration with the abundance of the genera Escherichia/Shigella or family Enterobacteriaceae among other nonoverlapping bacteria [122,124]. The abundances of Enterobacteriaceae also correlated with the severity of postural instability and gait disturbance in another independent study [126].
- Altered gut metabolites in PD
- The metabolites of gut microbiota, SCFAs, are produced by nondigestible carbohydrate fermentation via different metabolic pathways. The three most abundant SCFAs in the human colon are acetic acid, propionic acid, and butyric acid [136]. Different SCFA-producing bacteria adopt different metabolic pathways using different substrates and enzymes to selectively produce different SCFAs [136]. Many of these altered bacteria, e.g., from the families Prevotellaceae and Lachnospiraceae and genera Akkermansia, Blautia, Roseburia, and Faecalibacterium, comprise species that are SCFA producers and are decreased in PD patients except for the genus Akkermansia. One study also identified reduced fecal SCFA concentrations in PD patients [128].
- Accumulating evidence has suggested a role for SCFAs in maintaining gut barrier function and regulating gut motility and the immune response in the gut. One of the mechanisms for SCFAs in regulating the immune system in the gut is through gene expression control via histone deacetylase inhibition. This inhibition in colonocytes and immune cells by butyrate and propionate results in an increased expression of the anti-microbial cathelicidin-derived peptide of the innate immune system, IL-37, anti-inflammatory cytokines, IL-10, and transforming growth factor β [137]. It is also associated with the downregulation of proinflammatory cytokines, including IL-8, IL-6, IL-1β, IFN-γ, and TNF-α, and the promotion of colonic regulatory T cell differentiation to control intestinal inflammation [138]. Furthermore, butyrate, propionate, and acetate can interact with different G protein-coupled receptors (GPRs), including GPR41 (FFA3), GPR43 (FFA2), and GPR109A, located on the surface of host cells. There, they also exert an anti-inflammatory effect of T cell differentiation and modulation and NF-κB activation inhibition [138,139]. Despite evidence suggesting an intestinal health benefit of SCFAs, the major controversy for the role of gut microbiota in PD comes from a recent study showing that microbiota and SCFAs in the gut exacerbate α-synuclein pathology and microglia cell activation in α-synuclein–overexpressing mice [140]. Another detrimental effect of SCFAs, in particular propionic acid, was also shown to be related to other neurological disorders, such as autism, although the underlying mechanism remains unknown [141]. Different SCFAs have different effects on biological systems. To further delineate the role of SCFAs in PD, future studies should evaluate the individual SCFAs and their balance with each other instead of focusing on their effects as a whole.
- More research is needed to determine not only bacterial composition in PD patients but also changes in related biochemical pathways. These studies can rely on methods such as metabolomics, proteomics, metatranscriptomics, and shotgun metagenomic sequencing, which could lead to further insights into the role of altered gut microbiota and metabolites in the pathogenesis of PD.
- The effects of anti-PD medications on the gut microbiota
- One recent study found a significant difference in the gut microbiome as a function of treatment with COMT inhibitors and anticholinergics, and a borderline significance for carbidopa/levodopa [119]. As the growing literature on the role of the gut microbiome in the metabolism of medications and the profound effects that the drugs can have in turn on the composition of the microbiome [142-145], the interaction between use of PD medications and the compositions of gut microbiome is not surprising. Another prior study of PD also linked COMT inhibitors to altered abundance of some taxa [126]. Additionally, COMT inhibitors and anticholinergics have gastrointestinal side effects, which may contribute to altered gut microbiome [146,147]. These findings lend support to the notion that there is a clinically important relationship between microbiota and drug metabolism throughout the lifespan of PD patients; therefore, profiling of the human microbiome will be essential to understand the mechanisms by which these microbiota–drug interactions occur and the degree to which this complex interplay affects drug efficacy. Additional studies are needed to assess the potential utility of bacterial therapeutics in altering the microbiome to enhance therapeutic efficacy and clinical outcomes in patients with PD.
ALTERED GUT MICROBIOME IN PATIENTS WITH PD
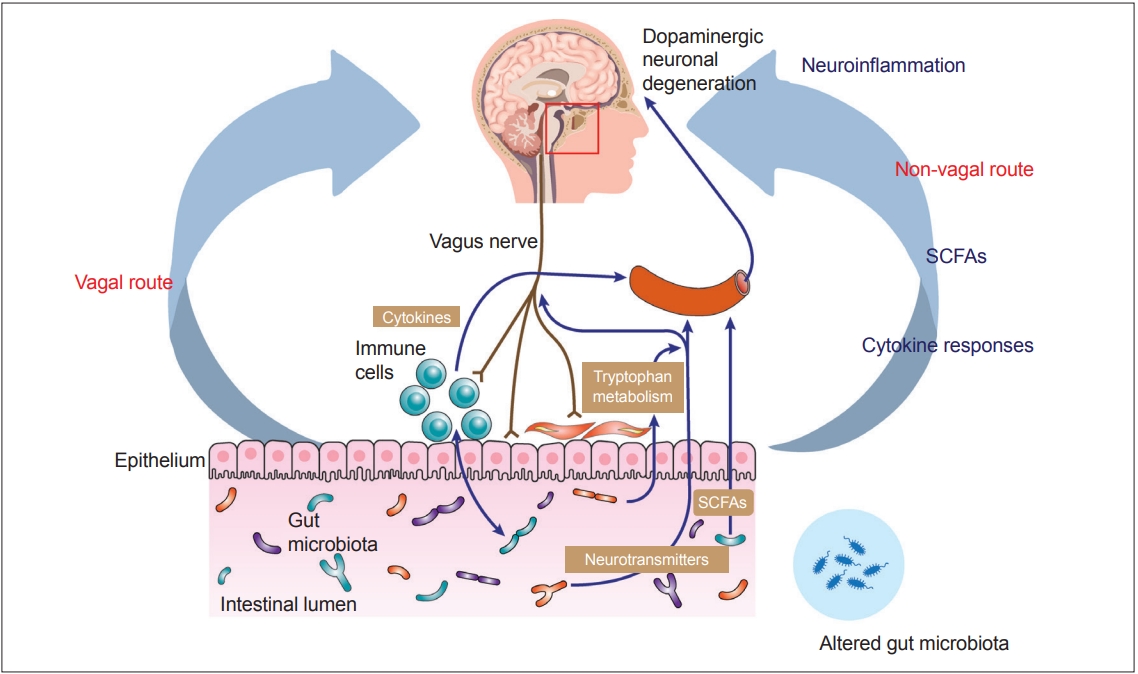
- Recent advances in understanding the gut–brain axis have linked microbiota and intestinal pathology to PD pathogenesis (see review [148]). Based on animal and clinical studies, changes in the composition of the gut microbiota are proposed to induce a pro-inflammatory response in the gut. According to this conceptualization, the inflammatory response increases gut permeability and exposure to endotoxins or other bacterial products and induces α-synuclein aggregations, which in turn propagate to the CNS via the vagus nerve. In addition, a systemic inflammatory response might disrupt the blood–brain barrier, which, in combination with α-synuclein delivery from the gut, further activates microglia and results in dopamine neuron degeneration (Figure 2) [12]. However, many pieces remain missing from this huge puzzle of the gut–brain axis in PD. Future investigations are necessary to define specific microbe-derived factors, immune effector functions, and microbiota–immune pathways for modulating gut–brain communications. These studies will reveal fundamental biological insights into PD pathogenesis, with the potential to inform the development of new microbial- and immune-based therapeutic strategies for treatment.
IMPLICATIONS FOR FUTURE GUT-TARGETED THERAPY FOR EARLY-STAGE PD
- The authors are grateful to the National Health Research Institutes (NHRI-EX107-10716NC) and the National Taiwan University Hospital (106-EDN06 and 107-EDN14) for their funding support of this work.
Acknowledgments

| Author (year) | Region | Anti-α-syn Ab (clone) | ENS involved | PD (positive/total number) | Control (positive/total number) | Presence of LB-like accumulations |
|---|---|---|---|---|---|---|
| Braak et al. (2006) [40] | Distal esophagus, stomach | Syn-1 (clone 42) | Au, Me, Adventitia | 5/5 | 0/5 | + (Au) |
| Bloch et al. (2006) [42] | Distal esophagus | α-syn (LB509) | Au > Me | 1/2 | 14/98 | N.A. |
| Beach et al. (2010) [46] | Esophagus, stomach, duodenum, ileum, colon | P-Ser129-α-syn | Au, Me | 11/17 | 0/23 | N.A. |
| Del Tredici et al. (2010) [43] | Distal esophagus, stomach | Syn-1 (clone 42) | N.A. | 8/8 | 1/13 | N.A. |
| Del Tredici et al. (2011) [148] | Distal esophagus, stomach | Syn-1 (clone 42) | Au | 3/3 | 1/1 | + (Au) |
| Annerino et al. (2012) [41]* | Stomach, duodenum, ileum, colon | α-syn (LB509) | Au > Me | 13/13 | 0/12 | + (Au > Me) |
| Gold et al. (2013) [45] | Colon | α-syn (KM51) | Au > Me | 10/10 | 52% | N.A. |
| Gelpi et al. (2014) [44]* | Esophagus, stomach, colon | α-syn (KM51) | Au | 8/10 | 5/5 (DLB), 0/8 (AD) | + (Au) |
| P-α-syn (pSyn#64) |
* Colocalization staining with neuronal markers was performed in only 2 studies [Annerino et al. (2012)[41] and Gelpi et al. (2014)[44]].
Ab: antibody, GI: gastrointestinal, PD: Parkinson’s disease, LB: Lewy body, DLB: diffuse Lewy body dementia, α-syn: alpha-synuclein, P-α-syn: phosphorylated α-synuclein, Au: Auerbach plexus, Me: Meissner plexus, N.A.: not available, AD: Alzheimer’s disease.
| Author (year) | Region | Depth | Method | α-synuclein Ab (clone or source) | Costaining neuronal marker | PD (positive/total number, or %) | Control (positive/total number, or %) |
|---|---|---|---|---|---|---|---|
| Lebouvier et al. (2008) [55] | Ascending colon | S | DIF (WM) | P-α-syn (pSyn#64) | Neurofilament, HuC/D, DBH | 4/5 | 0/5 |
| Lebouvier et al. (2010) [56] | Ascending and descending colon, | S | DIF (WM) | P-α-syn (pSyn#64) | Neurofilament, HuC/D, DBH | 20/29 | 0/10 |
| Shannon et al. (2012) [59] | Sigmoid | S | IHC | α-syn (LB509) | Cuprolinic blue solution | 9/9 | 2/24 |
| Shannon et al. (2012) [58] | Large intestine | S | IHC + DIF | α-syn (LB509) | Substance P | 3/3 | 0/23 |
| Pouclet et al. (2012) [57] | Ascending and descendin colon, rectum | S | DIF (WM) | P-α-syn (pSyn#64) | Neurofilament | Ascending colon 17/26; Descending colon 11/26; Rectum 6/26 | 0/9 |
| Pouclet et al. (2012) [53] | Descending colon, sigmoid | S | DIF (WM) | P-α-syn (pSyn#64) | Neurofilament | M 3/9; S 4/9 | 0/10 |
| Pouclet et al. (2012) [149] | Descending colon | S | DIF (WM) | P-α-syn (pSyn#64) | Neurofilament | 5/9 | MSA 1/6 |
| Hilton et al. (2014) [61] | Esophagus, stomach, small and large intestine | M + S | IHC + DIF | α-syn (KM51) | S-100 | 7/62 | 0/161 |
| P-α-syn (pSyn#64) | |||||||
| Sánchez-Ferro et al. (2015) [49] | Stomach | M | IHC | α-syn (KM51) | S-100 | 17/20 | 1/23 |
| P-α-syn (pSyn#64) | |||||||
| Visanji et al. (2015) [51] | Sigmoid, rectum | M | IHC, PET | α-syn (LB509) | - | α-syn 22/22 | α-syn 9/11 |
| P-α-syn (ab59264) | P-α-syn 22/22 | P-α-syn 10/11 | |||||
| Sprenger et al. (2015) [52] | Whole colon, sigmoid, rectum | M + S | IHC + DIF | α-syn (15G7) | HuC/HuD | α-syn: M 24/24; S 21/21 | α-syn: M 21/22; S16/16 |
| P-α-syn (pSyn#64) | P-α-syn: M 0/24; S 1/19 | P-α-syn: M 0/22; S 0/14 | |||||
| Clairembault et al. (2015) [54] | Descending colon, sigmoid | S | IHC + DIF | P-α-syn (pSyn#64) | PGP 9.5 | 23/31 | 0/11 |
| Chung et al. (2016) [47] | Stomach, large intestine | M | IHC | P-α-syn (EP1536Y) | S-100 | Stomach 31.6%; Colon 10.6% | Stomach 33.3%; Colon 18.5% |
| Mrabet et al. (2016) [62] | Esophagus, stomach, duodenum | M + S | IHC | α-syn (monoclonal antibody) | - | 30/30 | 1/13 |
| Stokholm et al. (2016) [65] | Esophagus, stomach, small and large intestine | S | IHC | α-syn (MJFR1) | Synaptophysin | α-syn: Pre-PD 24/39; PD 11/18 | α-syn 43/90 |
| P-α-syn (MJF-R13) | P-α-syn: Pre-PD 22/39; PD 9/18 | P-α-syn 23/90 | |||||
| Antunes et al. (2016) [50] | Large intestine | M | IHC | α-syn (Syn204) | - | α-syn 18/19 | α-syn 8/8 |
| P-α-syn (ab59264) | P-α-syn 19/19 | P-α-syn 8/8 | |||||
| Barrenschee et al. (2017) [60] | Rectum | M + S | DIF | P-α-syn (pSyn#64) | PGP 9.5 | 3/12 | 4/11 |
| Rouaud et al. (2017) [150] | Large intestine | S | IHC | P-α-syn (pSyn#64) | PGP 9.5 | 3/3 (LRRK2-G2019S) | - |
| Shin et al. (2017) [64] | Stomach, large intestine | M + S | IHC | P-α-syn (EP1536Y) | Neurofilament | 2/22 | 4/22 |
| Ruffmann et al. (2018) [63] | Esophagus, stomach, small and large intestine | M + S | IHC | α-syn (KM51, LB509) | Calretinin (5A5), Hu C/HuD | α-syn 0/51 | α-syn 0/21 |
| PET | O-α-syn | P-α-syn 7/51 | P-α-syn 5/21 | ||||
| P-α-syn (pSyn#64) | O-α-syn 10/51 | O-α-syn 5/21 | |||||
| Lee et al. (2018) [48] | Stomach, large intestine | M | IHC | P-α-syn (EP1536Y) | S-100 | 12/33 | 20/46 |
PD: Parkinson’s disease, Ab: antibody, S: submucosa, M: mucosa, IHC: immunohistochemistry, DIF: double immunofluorescence, WM: whole mount, PET: paraffin-embedded tissue blot, α-syn: α-synuclein, P-α-syn: phospho-α-synuclein, O-α-syn: oligomeric form of α-synuclein, T: total, TH: tyrosine hydroxylase, DBH: dopamine-beta-hydroxylase, MSA: multiple system atrophy, Pre-PD: premotor stage of PD, LRRK2-G2019S: PD patients carrying the G2019S mutation in LRRK2.
| Authors (year) | Region | Method | a-syn Ab (clone) | Costaining neuronal marker Ab | ENS involved | PD (positive/total number, or %) | Control (positive/total number, or %) |
|---|---|---|---|---|---|---|---|
| Minguez-Castellanos et al. (2007) [151] | Small and large intestine | IHC | a-syn (KM51) | PGP9.5, TH Neurofilament | N.A. | N.A. | 3.90% |
| a-syn (LB509) | |||||||
| Böttner et al. (2012) [69] | Colon, rectum | IHC + DIF | a-syn | HuC/D, PGP 9.5 | Au, Me | N.A. | a-syn: Au13/13, Me13/13 |
| P-a-syn (pSyn#64) | P-a-syn: Au11/13, Me 8/13 | ||||||
| Ito et al. (2014) [67] | Stomach, duodenum, small intestine, colon, gallbladder | IHC | P-a-syn (pSyn#64), P-a-syn (serine129, polyclonal) | Neurofilament, TH | Au > Me, Subserosa nerve fascicles | 2/2 | 0/10, 4/6 (DLB)* |
| Aldecoa et al. (2015) [66] | Stomach, small and large intestine | IHC | a-syn (KM51) | N.A. | Au > Me | 4/6 | 1/12 |
| P-a-syn (pSyn#64) | |||||||
| a-syn (15G7) | |||||||
| a-syn (505) | |||||||
| Shin et al. (2017) [64] | Stomach, large intestine | IHC | P-a-syn (EP1536Y) | Neurofilament | Au > Me | 12/33 | 6/33 |
| Yan et al. (2018) [68] | Stomach, intestine, appendix | IHC | a-syn (monoclonal antibody) | N.A. | Au, Me | 17/31 | 7/32 |
* 1 α-syn positive patient was preclinical.
ENS: enteric nervous system, PD: Parkinson’s disease, Ab: antibody, IHC: immunohistochemistry, DIF: double immunofluorescence, Au: Auerbach plexus, Me: Meissner plexus, DLB: diffuse Lewy body dementia, TH: tyrosine hydroxylase, α-syn: α-synuclein, P-α-syn: phospho-α-synuclein, N.A.: not available.
| Author (year) | Sequence region* | Pt/Ctrl | α/β diversity | Increased in PD | Decreased in PD |
|---|---|---|---|---|---|
| Scheperjans et al. (2015) [126] | V1, V2, V3 | 72/72 | α↔︎/β dif | Lactobacillaceae (F), Verrucomicrobiaceae (F), Bradyrhizobiaceae (F), Clostridiales (F), Incertae Sedis IV (F) | Prevotellaceae (F) |
| Keshavarzian et al. (2015) [121] | V4 | 38/34 | α↑/β dif | Bacteroidetes (P), Proteobacteria (P), Verrucomicrobia (P), Clostridiaceae (F), Oscillospira (G), Akkermansia (G) | Firmicutes (P), Lachnospiraceae (F), Coprobacillaceae (F), Blautia (G), Coprococcus (G), Dorea (G), Roseburia (G) |
| Hasegawa et al. (2015) [117] | Quantitative RT-PCR | 52/36 | - | Lactobacillus (G) | Species: Clostridium coccoides group, Clostridium leptum subgroup, Bacteroides fragilis group |
| Unger et al. (2016) [128] | Quantitative RT-PCR | 34/34 | - | Enterobacteriaceae (F), Bifidobacterium (G) | Bacteroidetes (P), Prevotellaceae (F, descriptively reduced), Lactobacillaceae (F), Enterococcaceae, Species: Faecalibacterium prausnitzii |
| Petrov et al. (2017) [124] | V3, V4 | 89/66 | α↓/β dif | Christensenella (G), Catabacter (G), Lactobacillus (G), Oscillospira (G), Bifidobacterium (G) | Dorea (G), Bacteroides (G), Prevotella (G), Faecalibacterium (G) |
| Bedarf et al. (2017) [116] | Metagenomic shotgun analysis | 31/28 | α↔︎/β dif | Verrucomicrobiaceae (F), Firmicutes (F), Akkermansia (G) | Prevotellaceae (F), Erysipelotrichaceae (F), Prevotella (G), Eubacterium (G) |
| Hill-Burns et al. (2017) [119] | N.A. | 197/130 | α↔︎/β dif | Bifidobacteriaceae (F), Lactobacillaceae (F), Tissierellaceae (F), Christensenellaceae (F), Verrucomicrobiaceae (F), Bifidobacterium (G), Lactobacillus (G), Akkermansia (G) | Lachnospiraceae (F), Pasteurellaceae (F), Blautia (G), Roseburia (G), Faecalibacterium (G) |
| Li et al. (2017) [122] | V3, V4, V5 | 24/14 | α↓/β dif | Actinobacteria (P), Proteobacteria (P), Enterobacteriaceae (F), Streptococcaceae (F), Veillonellaceae (F), Acidaminococcus (G), Acinetobacter (G), Enterococcus (G), Escherichia-Shigella (G), Megamonus (G), Megasphaera (G), Proteus (G), Streptococcus (G) | Bacteroidetes (P), Pasteurellaceae (F), Blautia (G), Faecalibacterium (G), Ruminococcus (G) |
| Hopfner et al. (2017) [120] | V1, V2 | 29/29 | α↔︎/β dif | Barnesiellaceae (F), Enterococcaceae (F), Lactobacillaceae (F) | - |
| Qian et al. (2018) [125] | V3, V4 | 45/45 | α↑/β dif | Clostridium IV (G), Aquabacterium (G), Holdemania (G), Sphingomonas (G), Clostridium XVIII (G), Butyricicoccus (G), Anaerotruncus (G) | - |
| Lin et al. (2018) [123] | V4 | 75/45 | α↔︎/β dif | Eubacteriaceae (F), Bifidobacteriaceae (F), Aerococcaceae (F), Desulfovibrionaceae (F) (see footnote for genera composition) | Tenericutes (P), Euryarchaeota (P), Firmicutes (P), Streptococcaceae (F), Methylobacteriaceae (F), Comamonadaceae (F), Halomonadaceae (F), Hyphomonadaceae (F), Brucellaceae (F), Xanthomonadaceae (F), Lachnospiraceae (F), Actinomycetaceae (F), Sphingomonadaceae (F), Pasteurellaceae (F), Micrococcaceae (F), Intrasporangiaceae (F), Methanobacteriaceae (F), Idiomarinaceae (F), Brevibacteriaceae (F), Gemellaceae (F) (see footnote for genera composition) |
| Heintz-Buschart et al. (2018) [118] | V4 | 76/78 (21 iRBD) | α↔︎/β dif | Verrucomicrobia (P), Verrucomicrobiales (O), Verrucomicrobiaceae (F), Akkermansia (G) | - |
| Tan et al. (2018) [127] | V3, V4 | 104/96 | α↔︎/β dif | - | Bacteroides (G), Parasutterella (G), Collinsella (G), Escherichia/Shigella (G), Prevotella (G), Bifidobacterium (G) |
Results from Lin et al. (2018)123: increased (genera) – Bifidobacterium, Morganella, Dialister, Desulfovibrio, Gardnerella, Ralstonia, Lachnobacterium, Holdemania, Alloiococcus, Alistipes, Bilophila; decreased (genera) – Eikenella, Ochrobactrum, Lachnospira, Bulleidia, Rubellimicrobium, Methylobacterium, Knoellia, Streptococcus, Succinatimonas, Dietzia, Hyphomonas, Lupinus, Brevibacterium, Hemophilus, Oenothera, Roseburia, Actinomyces, Blautia, Pedobacter, Pseudochrobacterum, Mycoplana, Faecalibacterium, Halomonas, Pseudobacterium, Metmaris, Stemonimonas, Stemonas, Stemonas, Metacuhingum, and Stemonas.
* 16S ribosomal RNA gene.
↔: similar, ↓: decrease, ↑: increase. PD: Parkinson’s disease, MSA: metagenomic shotgun analysis, (-): not available, Pt: patient, Ctrl: Control, dif: different, P: phylum, F: family, O: order, G: genus, iRBD: idiopathic REM sleep behavior disorder.
| Scheperjans et al. (2015) [126] | Keshavarzian et al. (2015) [121] | Hasegawa et al. (2015) [117] | Unger et al. (2016) [128] | Petrov et al. (2017) [124] | Bedarf et al. (2017) [116] | Hill-Burns et al. (2017) [119] | Li et al. (2017) [122] | Hopfner et al. (2017) [120] | Qian et al. (2018) [125] | Lin et al. (2018) [123] | Heintz-Buschart et al. (2018) [118] | Tan et al. (2018) [127] | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phylum | |||||||||||||
| Bacteroidetes | ↑ | ↓ | ↓ | ||||||||||
| Proteobacteria | ↑ | ↑ | |||||||||||
| Verrucomicrobia | ↑ | ↑ | |||||||||||
| Firmicutes | ↓ | ↑ | ↓ | ||||||||||
| Family | |||||||||||||
| Prevotellaceae | ↓ | ↓* | ↓ | ||||||||||
| Lactobacillaceae | ↑ | ↓ | ↑ | ↑ | |||||||||
| Verrucomicrobiaceae | ↑ | ↑ | ↑ | ↑ | |||||||||
| Lachnospiraceae | ↓ | ↓ | ↓ | ||||||||||
| Bifidobacteriaceae | ↑ | ↑ | |||||||||||
| Pasteurellaceae | ↓ | ↓ | ↓ | ||||||||||
| Enterobacteriaceae | ↑ | ↑ | |||||||||||
| Streptococcaceae | ↑ | ↓ | |||||||||||
| Enterococcaceae | ↓ | ↑ | |||||||||||
| Genus | |||||||||||||
| Oscillospira | ↑ | ↑ | |||||||||||
| Akkermansia | ↑ | ↑ | ↑ | ↑ | |||||||||
| Blautia | ↓ | ↓ | ↓ | ↓ | |||||||||
| Dorea | ↓ | ↓ | |||||||||||
| Roseburia | ↓ | ↓ | ↓ | ||||||||||
| Prevotella | ↓ | ↓ | ↓ | ||||||||||
| Bifidobacterium | ↑ | ↑ | ↑ | ↑ | ↓ | ||||||||
| Lactobacillus | ↑ | ↑ | ↑ | ||||||||||
| Faecalibacterium | ↓ | ↓ | |||||||||||
| Escherichia/shigella | ↑ | ↓ | |||||||||||
| Streptococcus | ↑ | ↓ | |||||||||||
| Faecalibacterium | ↓ | ↓ | ↓ | ||||||||||
| Holdemania | ↑ | ↑ | |||||||||||
| Bacteroides | ↓ | ↓ |
- 1. Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, et al. Parkinson disease. Nat Rev Dis Primers 2017;3:17013.ArticlePubMedPDF
- 2. Braak H, Rüb U, Gai WP, Del Tredici K. Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm (Vienna) 2003;110:517–536.ArticlePubMedPDF
- 3. Hawkes CH, Del Tredici K, Braak H. Parkinson’s disease: a dual-hit hypothesis. Neuropathol Appl Neurobiol 2007;33:599–614.ArticlePubMedPMC
- 4. Hawkes CH, Del Tredici K, Braak H. Parkinson’s disease: the dual hit theory revisited. Ann N Y Acad Sci 2009;1170:615–622.ArticlePubMed
- 5. Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat Rev Neurol 2013;9:13–24.ArticlePubMedPDF
- 6. Reichmann H. View point: etiology in Parkinson’s disease. Dual hit or spreading intoxication. J Neurol Sci 2011;310:9–11.ArticlePubMed
- 7. Chen SG, Stribinskis V, Rane MJ, Demuth DR, Gozal E, Roberts AM, et al. Exposure to the functional bacterial amyloid protein curli enhances alpha-synuclein aggregation in aged Fischer 344 rats and Caenorhabditis elegans. Sci Rep 2016;6:34477.ArticlePubMedPMCPDF
- 8. Kelly LP, Carvey PM, Keshavarzian A, Shannon KM, Shaikh M, Bakay RA, et al. Progression of intestinal permeability changes and alpha-synuclein expression in a mouse model of Parkinson’s disease. Mov Disord 2014;29:999–1009.ArticlePubMed
- 9. Pan-Montojo F, Anichtchik O, Dening Y, Knels L, Pursche S, Jung R, et al. Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS One 2010;5:e8762. ArticlePubMedPMC
- 10. Holmqvist S, Chutna O, Bousset L, Aldrin-Kirk P, Li W, Björklund T, et al. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol 2014;128:805–820.ArticlePubMedPDF
- 11. Ulusoy A, Phillips RJ, Helwig M, Klinkenberg M, Powley TL, Di Monte DA. Brain-to-stomach transfer of α-synuclein via vagal preganglionic projections. Acta Neuropathol 2017;133:381–393.ArticlePubMedPDF
- 12. Perez-Pardo P, Hartog M, Garssen J, Kraneveld AD. Microbes tickling your tummy: the importance of the gut-brain axis in Parkinson’s disease. Curr Behav Neurosci Rep 2017;4:361–368.ArticlePubMedPMCPDF
- 13. Attems J, Jellinger KA. The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson’s disease. Neuropathol Appl Neurobiol 2008;34:466–467.ArticlePubMed
- 14. Kalaitzakis ME, Graeber MB, Gentleman SM, Pearce RK. The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson’s disease: a critical analysis of alpha-synuclein staging. Neuropathol Appl Neurobiol 2008;34:284–295.ArticlePubMed
- 15. Liu B, Fang F, Pedersen NL, Tillander A, Ludvigsson JF, Ekbom A, et al. Vagotomy and Parkinson disease: a Swedish register-based matched-cohort study. Neurology 2017;88:1996–2002.ArticlePubMedPMC
- 16. Svensson E, Horváth-Puhó E, Thomsen RW, Djurhuus JC, Pedersen L, Borghammer P, et al. Vagotomy and subsequent risk of Parkinson’s disease. Ann Neurol 2015;78:522–529.ArticlePubMed
- 17. Tysnes OB, Kenborg L, Herlofson K, Steding-Jessen M, Horn A, Olsen JH, et al. Does vagotomy reduce the risk of Parkinson’s disease? Ann Neurol 2015;78:1011–1012.ArticlePubMed
- 18. Pfeiffer RF. Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol 2003;2:107–116.ArticlePubMed
- 19. Srivanitchapoom P, Pandey S, Hallett M. Drooling in Parkinson’s disease: a review. Parkinsonism Relat Disord 2014;20:1109–1118.ArticlePubMedPMC
- 20. Nicaretta DH, de Rosso AL, Maliska C, Costa MM. Scintigraphic analysis of the parotid glands in patients with sialorrhea and Parkinson’s disease. Parkinsonism Relat Disord 2008;14:338–341.ArticlePubMed
- 21. Kalf JG, Munneke M, van den Engel-Hoek L, de Swart BJ, Borm GF, Bloem BR, et al. Pathophysiology of diurnal drooling in Parkinson’s disease. Mov Disord 2011;26:1670–1676.ArticlePubMed
- 22. Pfeiffer RF. Gastrointestinal dysfunction in Parkinson’s disease. Curr Treat Options Neurol 2018;20:54.ArticlePubMedPDF
- 23. Fasano A, Visanji NP, Liu LW, Lang AE, Pfeiffer RF. Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol 2015;14:625–639.ArticlePubMed
- 24. Heetun ZS, Quigley EM. Gastroparesis and Parkinson’s disease: a systematic review. Parkinsonism Relat Disord 2012;18:433–440.ArticlePubMed
- 25. Doi H, Sakakibara R, Sato M, Masaka T, Kishi M, Tateno A, et al. Plasma levodopa peak delay and impaired gastric emptying in Parkinson’s disease. J Neurol Sci 2012;319:86–88.ArticlePubMed
- 26. Stocchi F, Torti M. Constipation in Parkinson’s disease. Int Rev Neurobiol 2017;134:811–826.ArticlePubMed
- 27. Knudsen K, Krogh K, Østergaard K, Borghammer P. Constipation in parkinson’s disease: subjective symptoms, objective markers, and new perspectives. Mov Disord 2017;32:94–105.ArticlePubMed
- 28. Edwards LL, Pfeiffer RF, Quigley EM, Hofman R, Balluff M. Gastrointestinal symptoms in Parkinson’s disease. Mov Disord 1991;6:151–156.ArticlePubMed
- 29. Abbott RD, Petrovitch H, White LR, Masaki KH, Tanner CM, Curb JD, et al. Frequency of bowel movements and the future risk of Parkinson’s disease. Neurology 2001;57:456–462.ArticlePubMed
- 30. Lin CH, Lin JW, Liu YC, Chang CH, Wu RM. Risk of Parkinson’s disease following severe constipation: a nationwide population-based cohort study. Parkinsonism Relat Disord 2014;20:1371–1375.ArticlePubMed
- 31. Savica R, Carlin JM, Grossardt BR, Bower JH, Ahlskog JE, Maraganore DM, et al. Medical records documentation of constipation preceding Parkinson disease: a case-control study. Neurology 2009;73:1752–1758.ArticlePubMedPMC
- 32. Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003;24:197–211.ArticlePubMed
- 33. Borghammer P. Is constipation in Parkinson’s disease caused by gut or brain pathology? Parkinsonism Relat Disord 2018;55:6–7.ArticlePubMed
- 34. Orimo S, Ghebremedhin E, Gelpi E. Peripheral and central autonomic nervous system: does the sympathetic or parasympathetic nervous system bear the brunt of the pathology during the course of sporadic PD? Cell Tissue Res 2018;373:267–286.ArticlePubMedPDF
- 35. Derkinderen P, Rouaud T, Lebouvier T, Bruley des Varannes S, Neunlist M, De Giorgio R. Parkinson disease: the enteric nervous system spills its guts. Neurology 2011;77:1761–1767.ArticlePubMed
- 36. Furness JB. The enteric nervous system and neurogastroenterology. Nat Rev Gastroenterol Hepatol 2012;9:286–294.ArticlePubMedPDF
- 37. Qualman SJ, Haupt HM, Yang P, Hamilton SR. Esophageal Lewy bodies associated with ganglion cell loss in achalasia. Similarity to Parkinson’s disease. Gastroenterology 1984;87:848–856.ArticlePubMed
- 38. Wakabayashi K, Takahashi H, Ohama E, Ikuta F. Parkinson’s disease: an immunohistochemical study of Lewy body-containing neurons in the enteric nervous system. Acta Neuropathol 1990;79:581–583.ArticlePubMedPDF
- 39. Wakabayashi K, Takahashi H, Takeda S, Ohama E, Ikuta F. Parkinson’s disease: the presence of Lewy bodies in Auerbach’s and Meissner’s plexuses. Acta Neuropathol 1988;76:217–221.ArticlePubMedPDF
- 40. Braak H, de Vos RA, Bohl J, Del Tredici K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett 2006;396:67–72.ArticlePubMed
- 41. Annerino DM, Arshad S, Taylor GM, Adler CH, Beach TG, Greene JG. Parkinson’s disease is not associated with gastrointestinal myenteric ganglion neuron loss. Acta Neuropathol 2012;124:665–680.ArticlePubMedPMCPDF
- 42. Bloch A, Probst A, Bissig H, Adams H, Tolnay M. Alpha-synuclein pathology of the spinal and peripheral autonomic nervous system in neurologically unimpaired elderly subjects. Neuropathol Appl Neurobiol 2006;32:284–295.ArticlePubMed
- 43. Del Tredici K, Hawkes CH, Ghebremedhin E, Braak H. Lewy pathology in the submandibular gland of individuals with incidental Lewy body disease and sporadic Parkinson’s disease. Acta Neuropathol 2010;119:703–713.ArticlePubMedPDF
- 44. Gelpi E, Navarro-Otano J, Tolosa E, Gaig C, Compta Y, Rey MJ, et al. Multiple organ involvement by alpha-synuclein pathology in Lewy body disorders. Mov Disord 2014;29:1010–1018.ArticlePubMed
- 45. Gold A, Turkalp ZT, Munoz DG. Enteric alpha-synuclein expression is increased in Parkinson’s disease but not Alzheimer’s disease. Mov Disord 2013;28:237–240.ArticlePubMed
- 46. Beach TG, Adler CH, Sue LI, Vedders L, Lue L, White Iii CL, et al. Multiorgan distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol 2010;119:689–702.ArticlePubMedPMCPDF
- 47. Chung SJ, Kim J, Lee HJ, Ryu HS, Kim K, Lee JH, et al. Alpha-synuclein in gastric and colonic mucosa in Parkinson’s disease: limited role as a biomarker. Mov Disord 2016;31:241–249.ArticlePubMed
- 48. Lee HJ, Jung KW, Chung SJ, Hong SM, Kim J, Lee JH, et al. Relation of enteric α-synuclein to gastrointestinal dysfunction in patients with Parkinson’s disease and in neurologically intact subjects. J Neurogastroenterol Motil 2018;24:469–478.ArticlePubMedPMCPDF
- 49. Sánchez-Ferro Á, Rábano A, Catalán MJ, Rodríguez-Valcárcel FC, Fernández Díez S, Herreros-Rodríguez J, et al. In vivo gastric detection of α-synuclein inclusions in Parkinson’s disease. Mov Disord 2015;30:517–524.ArticlePubMed
- 50. Antunes L, Frasquilho S, Ostaszewski M, Weber J, Longhino L, Antony P, et al. Similar alpha-synuclein staining in the colon mucosa in patients with Parkinson’s disease and controls. Mov Disord 2016;31:1567–1570.ArticlePubMed
- 51. Visanji NP, Marras C, Kern DS, Al Dakheel A, Gao A, Liu LW, et al. Colonic mucosal a-synuclein lacks specificity as a biomarker for Parkinson disease. Neurology 2015;84:609–616.ArticlePubMedPMC
- 52. Sprenger FS, Stefanova N, Gelpi E, Seppi K, Navarro-Otano J, Offner F, et al. Enteric nervous system alpha-synuclein immunoreactivity in idiopathic REM sleep behavior disorder. Neurology 2015;85:1761–1768.ArticlePubMedPMC
- 53. Pouclet H, Lebouvier T, Coron E, Des Varannes SB, Neunlist M, Derkinderen P. A comparison between colonic submucosa and mucosa to detect Lewy pathology in Parkinson’s disease. Neurogastroenterol Motil 2012;24:e202–205.ArticlePubMed
- 54. Clairembault T, Leclair-Visonneau L, Coron E, Bourreille A, Le Dily S, Vavasseur F, et al. Structural alterations of the intestinal epithelial barrier in Parkinson’s disease. Acta Neuropathol Commun 2015;3:12.ArticlePubMedPMCPDF
- 55. Lebouvier T, Chaumette T, Damier P, Coron E, Touchefeu Y, Vrignaud S, et al. Pathological lesions in colonic biopsies during Parkinson’s disease. Gut 2008;57:1741–1743.ArticlePubMedPMC
- 56. Lebouvier T, Neunlist M, Bruley des Varannes S, Coron E, Drouard A, N’Guyen JM, et al. Colonic biopsies to assess the neuropathology of Parkinson’s disease and its relationship with symptoms. PLoS One 2010;5:e12728. ArticlePubMedPMC
- 57. Pouclet H, Lebouvier T, Coron E, des Varannes SB, Rouaud T, Roy M, et al. A comparison between rectal and colonic biopsies to detect Lewy pathology in Parkinson’s disease. Neurobiol Dis 2012;45:305–309.ArticlePubMed
- 58. Shannon KM, Keshavarzian A, Dodiya HB, Jakate S, Kordower JH. Is alpha-synuclein in the colon a biomarker for premotor Parkinson’s disease? Evidence from 3 cases. Mov Disord 2012;27:716–719.ArticlePubMed
- 59. Shannon KM, Keshavarzian A, Mutlu E, Dodiya HB, Daian D, Jaglin JA, et al. Alpha-synuclein in colonic submucosa in early untreated Parkinson’s disease. Mov Disord 2012;27:709–715.ArticlePubMedPMC
- 60. Barrenschee M, Zorenkov D, Böttner M, Lange C, Cossais F, Scharf AB, et al. Distinct pattern of enteric phospho-alpha-synuclein aggregates and gene expression profiles in patients with Parkinson’s disease. Acta Neuropathol Commun 2017;5:1.ArticlePubMedPMCPDF
- 61. Hilton D, Stephens M, Kirk L, Edwards P, Potter R, Zajicek J, et al. Accumulation of alpha-synuclein in the bowel of patients in the pre-clinical phase of Parkinson’s disease. Acta Neuropathol 2014;127:235–241.ArticlePubMedPMCPDF
- 62. Mrabet S, Ben Ali N, Achouri A, Dabbeche R, Najjar T, Haouet S, et al. Gastrointestinal dysfunction and neuropathologic correlations in Parkinson disease. J Clin Gastroenterol 2016;50:e85–e90.ArticlePubMed
- 63. Ruffmann C, Bengoa-Vergniory N, Poggiolini I, Ritchie D, Hu MT, Alegre-Abarrategui J, et al. Detection of alpha-synuclein conformational variants from gastro-intestinal biopsy tissue as a potential biomarker for Parkinson’s disease. Neuropathol Appl Neurobiol 2018;44:722–736.ArticlePubMedPMC
- 64. Shin C, Park SH, Yun JY, Shin JH, Yang HK, Lee HJ, et al. Fundamental limit of alpha-synuclein pathology in gastrointestinal biopsy as a pathologic biomarker of Parkinson’s disease: comparison with surgical specimens. Parkinsonism Relat Disord 2017;44:73–78.ArticlePubMed
- 65. Stokholm MG, Danielsen EH, Hamilton-Dutoit SJ, Borghammer P. Pathological alpha-synuclein in gastrointestinal tissues from prodromal Parkinson disease patients. Ann Neurol 2016;79:940–949.ArticlePubMed
- 66. Aldecoa I, Navarro-Otano J, Stefanova N, Sprenger FS, Seppi K, Poewe W, et al. Alpha-synuclein immunoreactivity patterns in the enteric nervous system. Neurosci Lett 2015;602:145–149.ArticlePubMed
- 67. Ito S, Takao M, Hatsuta H, Kanemaru K, Arai T, Saito Y, et al. Alphasynuclein immunohistochemistry of gastrointestinal and biliary surgical specimens for diagnosis of Lewy body disease. Int J Clin Exp Pathol 2014;7:1714–1723.PubMedPMC
- 68. Yan F, Chen Y, Li M, Wang Y, Zhang W, Chen X, et al. Gastrointestinal nervous system α-synuclein as a potential biomarker of Parkinson disease. Medicine (Baltimore) 2018;97:e11337. ArticlePubMedPMC
- 69. Böttner M, Zorenkov D, Hellwig I, Barrenschee M, Harde J, Fricke T, et al. Expression pattern and localization of alpha-synuclein in the human enteric nervous system. Neurobiol Dis 2012;48:474–480.ArticlePubMed
- 70. Gray MT, Munoz DG, Gray DA, Schlossmacher MG, Woulfe JM. Alphasynuclein in the appendiceal mucosa of neurologically intact subjects. Mov Disord 2014;29:991–998.ArticlePubMed
- 71. Corbillé AG, Letournel F, Kordower JH, Lee J, Shanes E, Neunlist M, et al. Evaluation of alpha-synuclein immunohistochemical methods for the detection of Lewy-type synucleinopathy in gastrointestinal biopsies. Acta Neuropathol Commun 2016;4:35.ArticlePubMedPMC
- 72. Beach TG, Corbillé AG, Letournel F, Kordower JH, Kremer T, Munoz DG, et al. Multicenter assessment of immunohistochemical methods for pathological alpha-synuclein in sigmoid colon of autopsied Parkinson’s disease and control subjects. J Parkinsons Dis 2016;6:761–770.ArticlePubMedPMC
- 73. Punsoni M, Friedman JH, Resnick M, Donahue JE, Yang DF, Stopa EG. Enteric pathologic manifestations of alpha-synucleinopathies. Appl Immunohistochem Mol Morphol 2017;Nov. 11. [Epub]. http://dx.doi.org/10.1097/PAI.0000000000000613. Article
- 74. Kupsky WJ, Grimes MM, Sweeting J, Bertsch R, Cote LJ. Parkinson’s disease and megacolon: concentric hyaline inclusions (Lewy bodies) in enteric ganglion cells. Neurology 1987;37:1253–1255.ArticlePubMed
- 75. Corbillé AG, Coron E, Neunlist M, Derkinderen P, Lebouvier T. Appraisal of the dopaminergic and noradrenergic innervation of the submucosal plexus in PD. J Parkinsons Dis 2014;4:571–576.ArticlePubMed
- 76. Clairembault T, Leclair-Visonneau L, Neunlist M, Derkinderen P. Enteric glial cells: new players in Parkinson’s disease? Mov Disord 2015;30:494–498.ArticlePubMed
- 77. Clairembault T, Kamphuis W, Leclair-Visonneau L, Rolli-Derkinderen M, Coron E, Neunlist M, et al. Enteric GFAP expression and phosphorylation in Parkinson’s disease. J Neurochem 2014;130:805–815.ArticlePubMed
- 78. Devos D, Lebouvier T, Lardeux B, Biraud M, Rouaud T, Pouclet H, et al. Colonic inflammation in Parkinson’s disease. Neurobiol Dis 2013;50:42–48.ArticlePubMed
- 79. Villarán RF, Espinosa-Oliva AM, Sarmiento M, De Pablos RM, Argüelles S, Delgado-Cortés MJ, et al. Ulcerative colitis exacerbates lipopolysaccharide-induced damage to the nigral dopaminergic system: potential risk factor in Parkinson`s disease. J Neurochem 2010;114:1687–1700.ArticlePubMed
- 80. Houser MC, Chang J, Factor SA, Molho ES, Zabetian CP, Hill-Burns EM, et al. Stool immune profiles evince gastrointestinal inflammation in Parkinson’s disease. Mov Disord 2018;33:793–804.ArticlePubMedPMC
- 81. Schwiertz A, Spiegel J, Dillmann U, Grundmann D, Bürmann J, Faβbender K, et al. Fecal markers of intestinal inflammation and intestinal permeability are elevated in Parkinson’s disease. Parkinsonism Relat Disord 2018;50:104–107.ArticlePubMedPMC
- 82. Forsyth CB, Shannon KM, Kordower JH, Voigt RM, Shaikh M, Jaglin JA, et al. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLoS One 2011;6:e28032. ArticlePubMedPMC
- 83. Salat-Foix D, Tran K, Ranawaya R, Meddings J, Suchowersky O. Increased intestinal permeability and Parkinson disease patients: chicken or egg? Can J Neurol Sci 2012;39:185–188.ArticlePubMed
- 84. Hui KY, Fernandez-Hernandez H, Hu J, Schaffner A, Pankratz N, Hsu NY, et al. Functional variants in the LRRK2 gene confer shared effects on risk for Crohn’s disease and Parkinson’s disease. Sci Transl Med 2018;10:eaai7795.ArticlePubMedPMC
- 85. Bae JR, Lee BD. Function and dysfunction of leucine-rich repeat kinase 2 (LRRK2): Parkinson’s disease and beyond. BMB Rep 2015;48:243–248.ArticlePubMedPMCPDF
- 86. Lin JC, Lin CS, Hsu CW, Lin CL, Kao CH. Association between Parkinson’s disease and inflammatory bowel disease: a nationwide Taiwanese retrospective cohort study. Inflamm Bowel Dis 2016;22:1049–1055.ArticlePubMed
- 87. Villumsen M, Aznar S, Pakkenberg B, Jess T, Brudek T. Inflammatory bowel disease increases the risk of Parkinson’s disease: a Danish nationwide cohort study 1977-2014. Gut 2019;68:18–24.ArticlePubMed
- 88. Peter I, Dubinsky M, Bressman S, Park A, Lu C, Chen N, et al. Anti-tumor necrosis factor therapy and incidence of Parkinson disease among patients with inflammatory bowel disease. JAMA Neurol 2018;75:939–946.ArticlePubMedPMC
- 89. Weimers P, Halfvarson J, Sachs MC, Saunders-Pullman R, Ludvigsson JF, Peter I, et al. Inflammatory bowel disease and Parkinson’s disease: a nationwide Swedish cohort study. Inflamm Bowel Dis 2019;25:111–123.ArticlePubMedPDF
- 90. Camacho-Soto A, Gross A, Searles Nielsen S, Dey N, Racette BA. Inflammatory bowel disease and risk of Parkinson’s disease in Medicare beneficiaries. Parkinsonism Relat Disord 2018;50:23–28.ArticlePubMedPMC
- 91. Fujioka S, Curry SE, Kennelly KD, Tacik P, Heckman MG, Tsuboi Y, et al. Occurrence of Crohn’s disease with Parkinson’s disease. Parkinsonism Relat Disord 2017;37:116–117.ArticlePubMedPMC
- 92. Blaecher C, Smet A, Flahou B, Pasmans F, Ducatelle R, Taylor D, et al. Significantly higher frequency of Helicobacter suis in patients with idiopathic parkinsonism than in control patients. Aliment Pharmacol Ther 2013;38:1347–1353.ArticlePubMedPMC
- 93. Charlett A, Dobbs RJ, Dobbs SM, Weller C, Brady P, Peterson DW. Parkinsonism: siblings share Helicobacter pylori seropositivity and facets of syndrome. Acta Neurol Scand 1999;99:26–35.ArticlePubMed
- 94. Charlett A, Dobbs RJ, Dobbs SM, Weller C, Ibrahim MA, Dew T, et al. Blood profile holds clues to role of infection in a premonitory state for idiopathic parkinsonism and of gastrointestinal infection in established disease. Gut Pathog 2009;1:20.ArticlePubMedPMC
- 95. Dobbs SM, Dobbs RJ, Weller C, Charlett A, Bjarnason IT, Lawson AJ, et al. Differential effect of Helicobacter pylori eradication on time-trends in brady/hypokinesia and rigidity in idiopathic parkinsonism. Helicobacter 2010;15:279–294.ArticlePubMedPMC
- 96. Efthymiou G, Dardiotis E, Liaskos C, Marou E, Tsimourtou V, Rigopoulou EI, et al. Immune responses against Helicobacter pylori-specific antigens differentiate relapsing remitting from secondary progressive multiple sclerosis. Sci Rep 2017;7:7929.ArticlePubMedPMCPDF
- 97. Fasano A, Bove F, Gabrielli M, Petracca M, Zocco MA, Ragazzoni E, et al. The role of small intestinal bacterial overgrowth in Parkinson’s disease. Mov Disord 2013;28:1241–1249.ArticlePubMed
- 98. Nafisah W, Najman A, Hamizah R, Azmin S, Rabani R, Shah SA. High prevalence of Helicobacter pylori infection in Malaysian Parkinson’s disease patients. Journal of Parkinsonism and Restless Legs Syndrome 2013;3:63–67.Article
- 99. Nielsen HH, Qiu J, Friis S, Wermuth L, Ritz B. Treatment for Helicobacter pylori infection and risk of Parkinson’s disease in Denmark. Eur J Neurol 2012;19:864–869.ArticlePubMedPMC
- 100. Tsolaki F, Kountouras J, Topouzis F, Tsolaki M. Helicobacter pylori infection, dementia and primary open-angle glaucoma: are they connected? BMC Ophthalmol 2015;15:24.ArticlePubMedPMCPDF
- 101. Bu XL, Wang X, Xiang Y, Shen LL, Wang QH, Liu YH, et al. The association between infectious burden and Parkinson’s disease: a case-control study. Parkinsonism Relat Disord 2015;21:877–881.ArticlePubMed
- 102. Dardiotis E, Tsouris Z, Mentis AA, Siokas V, Michalopoulou A, Sokratous M, et al. H. pylori and Parkinson’s disease: meta-analyses including clinical severity. Clin Neurol Neurosurg 2018;175:16–24.ArticlePubMed
- 103. Hashim H, Azmin S, Razlan H, Yahya NW, Tan HJ, Manaf MR, et al. Eradication of Helicobacter pylori infection improves levodopa action, clinical symptoms and quality of life in patients with Parkinson’s disease. PLoS One 2014;9:e112330. ArticlePubMedPMC
- 104. Lee WY, Yoon WT, Shin HY, Jeon SH, Rhee PL. Helicobacter pylori infection and motor fluctuations in patients with Parkinson’s disease. Mov Disord 2008;23:1696–1700.ArticlePubMed
- 105. Mridula KR, Borgohain R, Chandrasekhar Reddy V, Bandaru VCh, Suryaprabha T. Association of Helicobacter pylori with Parkinson’s disease. J Clin Neurol 2017;13:181–186.ArticlePubMedPMC
- 106. Narożańska E, Białecka M, Adamiak-Giera U, Gawrońska-Szklarz B, Sołtan W, Schinwelski M, et al. Pharmacokinetics of levodopa in patients with Parkinson disease and motor fluctuations depending on the presence of Helicobacter pylori infection. Clin Neuropharmacol 2014;37:96–99.ArticlePubMed
- 107. Tan AH, Mahadeva S, Marras C, Thalha AM, Kiew CK, Yeat CM, et al. Helicobacter pylori infection is associated with worse severity of Parkinson’s disease. Parkinsonism Relat Disord 2015;21:221–225.ArticlePubMed
- 108. Liu H, Su W, Li S, Du W, Ma X, Jin Y, et al. Eradication of Helicobacter pylori infection might improve clinical status of patients with Parkinson’s disease, especially on bradykinesia. Clin Neurol Neurosurg 2017;160:101–104.ArticlePubMed
- 109. Pierantozzi M, Pietroiusti A, Galante A, Sancesario G, Lunardi G, Fedele E, et al. Helicobacter pylori-induced reduction of acute levodopa absorption in Parkinson’s disease patients. Ann Neurol 2001;50:686–687.ArticlePubMed
- 110. McGee DJ, Lu XH, Disbrow EA. Stomaching the possibility of a pathogenic role for Helicobacter pylori in Parkinson’s disease. J Parkinsons Dis 2018;8:367–374.ArticlePubMedPMC
- 111. DiBaise JK, Crowell MD, Driver-Dunckley E, Mehta SH, Hoffman-Snyder C, Lin T, et al. Weight loss in Parkinson’s disease: no evidence for role of small intestinal bacterial overgrowth. J Parkinsons Dis 2018;8:571–581.PubMed
- 112. Gabrielli M, Bonazzi P, Scarpellini E, Bendia E, Lauritano EC, Fasano A, et al. Prevalence of small intestinal bacterial overgrowth in Parkinson’s disease. Mov Disord 2011;26:889–892.ArticlePubMed
- 113. Niu XL, Liu L, Song ZX, Li Q, Wang ZH, Zhang JL, et al. Prevalence of small intestinal bacterial overgrowth in Chinese patients with Parkinson’s disease. J Neural Transm (Vienna) 2016;123:1381–1386.ArticlePubMedPDF
- 114. Tan AH, Mahadeva S, Thalha AM, Gibson PR, Kiew CK, Yeat CM, et al. Small intestinal bacterial overgrowth in Parkinson’s disease. Parkinsonism Relat Disord 2014;20:535–540.ArticlePubMed
- 115. Khoshini R, Dai SC, Lezcano S, Pimentel M. A systematic review of diagnostic tests for small intestinal bacterial overgrowth. Dig Dis Sci 2008;53:1443–1454.ArticlePubMedPDF
- 116. Bedarf JR, Hildebrand F, Coelho LP, Sunagawa S, Bahram M, Goeser F, et al. Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naïve Parkinson’s disease patients. Genome Med 2017;9:39.ArticlePubMedPMCPDF
- 117. Hasegawa S, Goto S, Tsuji H, Okuno T, Asahara T, Nomoto K, et al. Intestinal dysbiosis and lowered serum lipopolysaccharide-binding protein in Parkinson’s disease. PLoS One 2015;10:e0142164. ArticlePubMedPMC
- 118. Heintz-Buschart A, Pandey U, Wicke T, Sixel-Döring F, Janzen A, Sittig-Wiegand E, et al. The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eye movement sleep behavior disorder. Mov Disord 2018;33:88–98.ArticlePubMed
- 119. Hill-Burns EM, Debelius JW, Morton JT, Wissemann WT, Lewis MR, Wallen ZD, et al. Parkinson’s disease and Parkinson’s disease medications have distinct signatures of the gut microbiome. Mov Disord 2017;32:739–749.ArticlePubMedPMC
- 120. Hopfner F, Künstner A, Müller SH, Künzel S, Zeuner KE, Margraf NG, et al. Gut microbiota in Parkinson disease in a northern German cohort. Brain Res 2017;1667:41–45.ArticlePubMed
- 121. Keshavarzian A, Green SJ, Engen PA, Voigt RM, Naqib A, Forsyth CB, et al. Colonic bacterial composition in Parkinson’s disease. Mov Disord 2015;30:1351–1360.ArticlePubMed
- 122. Li W, Wu X, Hu X, Wang T, Liang S, Duan Y, et al. Structural changes of gut microbiota in Parkinson’s disease and its correlation with clinical features. Sci China Life Sci 2017;60:1223–1233.ArticlePubMedPDF
- 123. Lin A, Zheng W, He Y, Tang W, Wei X, He R, et al. Gut microbiota in patients with Parkinson’s disease in southern China. Parkinsonism Relat Disord 2018;53:82–88.ArticlePubMed
- 124. Petrov VA, Saltykova IV, Zhukova IA, Alifirova VM, Zhukova NG, Dorofeeva YB, et al. Analysis of gut microbiota in patients with Parkinson’s disease. Bull Exp Biol Med 2017;162:734–737.ArticlePubMedPDF
- 125. Qian Y, Yang X, Xu S, Wu C, Song Y, Qin N, et al. Alteration of the fecal microbiota in Chinese patients with Parkinson’s disease. Brain Behav Immun 2018;70:194–202.ArticlePubMed
- 126. Scheperjans F, Aho V, Pereira PA, Koskinen K, Paulin L, Pekkonen E, et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov Disord 2015;30:350–358.ArticlePubMed
- 127. Tan AH, Chong CW, Teh CSJ, Yap IKS, Loke MF, Bowman J, et al. Unveiling the function of altered gut microbiota composition in Parkinson’s disease. Mov Disord 2018;33:S783–S784.Article
- 128. Unger MM, Spiegel J, Dillmann KU, Grundmann D, Philippeit H, Bürmann J, et al. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat Disord 2016;32:66–72.ArticlePubMed
- 129. Ley RE. Gut microbiota in 2015: prevotella in the gut: choose carefully. Nat Rev Gastroenterol Hepatol 2016;13:69–70.ArticlePubMedPDF
- 130. Brown CT, Davis-Richardson AG, Giongo A, Gano KA, Crabb DB, Mukherjee N, et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS One 2011;6:e25792. ArticlePubMedPMC
- 131. Ganesh BP, Klopfleisch R, Loh G, Blaut M. Commensal akkermansia muciniphila exacerbates gut inflammation in salmonella typhimurium-infected gnotobiotic mice. PLoS One 2013;8:e74963. ArticlePubMedPMC
- 132. Miquel S, Martín R, Rossi O, Bermúdez-Humarán LG, Chatel JM, Sokol H, et al. Faecalibacterium prausnitzii and human intestinal health. Curr Opin Microbiol 2013;16:255–261.ArticlePubMed
- 133. Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A 2008;105:16731–16736.ArticlePubMedPMC
- 134. Martín R, Chain F, Miquel S, Lu J, Gratadoux JJ, Sokol H, et al. The commensal bacterium Faecalibacterium prausnitzii is protective in DNBS-induced chronic moderate and severe colitis models. Inflamm Bowel Dis 2014;20:417–430.ArticlePubMed
- 135. Berry D, Reinisch W. Intestinal microbiota: a source of novel biomarkers in inflammatory bowel diseases? Best Pract Res Clin Gastroenterol 2013;27:47–58.ArticlePubMed
- 136. Ríos-Covián D, Ruas-Madiedo P, Margolles A, Gueimonde M, de Los Reyes-Gavilan CG, Salazar N. Intestinal short chain fatty acids and their link with diet and human health. Front Microbiol 2016;7:185.ArticlePubMedPMC
- 137. Canani RB, Costanzo MD, Leone L, Pedata M, Meli R, Calignano A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J Gastroenterol 2011;17:1519–1528.ArticlePubMedPMC
- 138. Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol 2014;12:661–672.ArticlePubMedPDF
- 139. Sun M, Wu W, Liu Z, Cong Y. Microbiota metabolite short chain fatty acids, GPCR, and inflammatory bowel diseases. J Gastroenterol 2017;52:1–8.ArticlePubMedPDF
- 140. Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 2016;167:1469–1480.ArticlePubMedPMC
- 141. Mulak A. A controversy on the role of short-chain fatty acids in the pathogenesis of Parkinson’s disease. Mov Disord 2018;33:398–401.ArticlePubMed
- 142. Falony G, Joossens M, Vieira-Silva S, Wang J, Darzi Y, Faust K, et al. Population-level analysis of gut microbiome variation. Science 2016;352:560–564.ArticlePubMed
- 143. Zhernakova A, Kurilshikov A, Bonder MJ, Tigchelaar EF, Schirmer M, Vatanen T, et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 2016;352:565–569.ArticlePubMedPMC
- 144. Wilson ID, Nicholson JK. Gut microbiome interactions with drug metabolism, efficacy, and toxicity. Transl Res 2017;179:204–222.ArticlePubMed
- 145. Wilkinson EM, Ilhan ZE, Herbst-Kralovetz MM. Microbiota-drug interactions: impact on metabolism and efficacy of therapeutics. Maturitas 2018;112:53–63.ArticlePubMed
- 146. Kaakkola S. Clinical pharmacology, therapeutic use and potential of COMT inhibitors in Parkinson’s disease. Drugs 2000;59:1233–1250.ArticlePubMed
- 147. Ness J, Hoth A, Barnett MJ, Shorr RI, Kaboli PJ. Anticholinergic medications in community-dwelling older veterans: prevalence of anticholinergic symptoms, symptom burden, and adverse drug events. Am J Geriatr Pharmacother 2006;4:42–51.ArticlePubMed
- 148. Del Tredici K, Duda JE. Peripheral Lewy body pathology in Parkinson’s disease and incidental Lewy body disease: four cases. J Neurol Sci 2011;310:100–106.ArticlePubMed
- 149. Pouclet H, Lebouvier T, Coron E, Rouaud T, Flamant M, Toulgoat F, et al. Analysis of colonic alpha-synuclein pathology in multiple system atrophy. Parkinsonism Relat Disord 2012;18:893–895.ArticlePubMed
- 150. Rouaud T, Clairembault T, Coron E, Neunlist M, Anheim M, Derkinderen P. Enteric alpha-synuclein pathology in LRRK2-G2019S Parkinson’s disease. Parkinsonism Relat Disord 2017;40:83–84.ArticlePubMed
- 151. Minguez-Castellanos A, Chamorro CE, Escamilla-Sevilla F, Ortega-Moreno A, Rebollo AC, Gomez-Rio M, et al. Do alpha-synuclein aggregates in autonomic plexuses predate Lewy body disorders?: a cohort study. Neurology 2007;68:2012–2018.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
- Adipose tissue, systematic inflammation, and neurodegenerative diseases
AnaPaula de A. Boleti, PedroHenrique de O. Cardoso, BrenoEmanuel F. Frihling, PatríciaSouza e Silva, LuizFilipe R. N. de Moraes, Ludovico Migliolo
Neural Regeneration Research.2023; 18(1): 38. CrossRef - Parkinson's Disease: A Multisystem Disorder
Helena Nunes Costa, Ana Raquel Esteves, Nuno Empadinhas, Sandra Morais Cardoso
Neuroscience Bulletin.2023; 39(1): 113. CrossRef - Brain modulation by the gut microbiota: From disease to therapy
Sarmistha Mitra, Raju Dash, Amena Al Nishan, Sarmin Ummey Habiba, Il Soo Moon
Journal of Advanced Research.2023; 53: 153. CrossRef - Plasma Metabolic Analysis Reveals the Dysregulation of Short-Chain Fatty Acid Metabolism in Parkinson’s Disease
Ao Qi, Lulu Liu, Junjie Zhang, Simei Chen, Simin Xu, Yusen Chen, Lijiang Zhang, Chun Cai
Molecular Neurobiology.2023; 60(5): 2619. CrossRef - Fecal microbiota transplantation in Parkinson's disease—A randomized repeat-dose, placebo-controlled clinical pilot study
Herbert L. DuPont, Jessika Suescun, Zhi-Dong Jiang, Eric L. Brown, Heather T. Essigmann, Ashley S. Alexander, Andrew W. DuPont, Tehseen Iqbal, Netanya S. Utay, Michael Newmark, Mya C. Schiess
Frontiers in Neurology.2023;[Epub] CrossRef - Mesoporous silica nanoparticles: Their potential as drug delivery carriers and nanoscavengers in Alzheimer's and Parkinson's diseases
Mohamed S. Attia, Ahmed Yahya, Nada Abdel Monaem, Shereen A. Sabry
Saudi Pharmaceutical Journal.2023; 31(3): 417. CrossRef - GSDMD in peripheral myeloid cells regulates microglial immune training and neuroinflammation in Parkinson's disease
Bingwei Wang, Yan Ma, Sheng Li, Hang Yao, Mingna Gu, Ying Liu, You Xue, Jianhua Ding, Chunmei Ma, Shuo Yang, Gang Hu
Acta Pharmaceutica Sinica B.2023; 13(6): 2663. CrossRef - Gut-microbiome-brain axis: the crosstalk between the vagus nerve, alpha-synuclein and the brain in Parkinson’s disease
Júlio César Claudino dos Santos, Leandro Freitas Oliveira, Felipe Micelli Noleto, Camilla Teixeira Pinheiro Gusmão, Gerly Anne de Castro Brito, Glauce Socorro de Barros Viana
Neural Regeneration Research.2023; 18(12): 2611. CrossRef - Beyond the Microbiota: Understanding the Role of the Enteric Nervous System in Parkinson’s Disease from Mice to Human
Martina Montanari, Paola Imbriani, Paola Bonsi, Giuseppina Martella, Antonella Peppe
Biomedicines.2023; 11(6): 1560. CrossRef - Effects of gut microbiota on neurodegenerative diseases
Saima Khatoon, Nida Kalam, Summya Rashid, Gulnaz Bano
Frontiers in Aging Neuroscience.2023;[Epub] CrossRef - From-Toilet-to-Freezer: A Review on Requirements for an Automatic Protocol to Collect and Store Human Fecal Samples for Research Purposes
Frances Widjaja, Ivonne M. C. M. Rietjens
Biomedicines.2023; 11(10): 2658. CrossRef - Association between the risk and severity of Parkinson’s disease and plasma homocysteine, vitamin B12 and folate levels: a systematic review and meta-analysis
Yuxin Quan, Jisen Xu, Qing Xu, Zhiqing Guo, Ruwei Ou, Huifang Shang, Qianqian Wei
Frontiers in Aging Neuroscience.2023;[Epub] CrossRef - Oral pathogens exacerbate Parkinson’s disease by promoting Th1 cell infiltration in mice
Xue-Bing Bai, Shuo Xu, Lu-Jun Zhou, Xiao-Qian Meng, Yu-Lin Li, Yan-Lin Chen, Yi-Han Jiang, Wen-Zhen Lin, Bo-Yan Chen, Lin-Juan Du, Guo-Cai Tian, Yan Liu, Sheng-Zhong Duan, Ya-Qin Zhu
Microbiome.2023;[Epub] CrossRef - The intestinal luminal sources of α-synuclein: a gastroenterologist perspective
Aaron Lerner
Nutrition Reviews.2022; 80(2): 282. CrossRef - Mild Chronic Colitis Triggers Parkinsonism in LRRK2 Mutant Mice Through Activating TNF‐α Pathway
Chin‐Hsien Lin, Han‐Yi Lin, En‐Pong Ho, Yi‐Ci Ke, Mei‐Fang Cheng, Chyng‐Yann Shiue, Chi‐Han Wu, Peng‐Hsiang Liao, Angela Yu‐Huey Hsu, Li‐An Chu, Ya‐Ding Liu, Ya‐Hui Lin, Yi‐Cheng Tai, Chia‐Tung Shun, Han‐Mo Chiu, Ming‐Shiang Wu
Movement Disorders.2022; 37(4): 745. CrossRef - Digesting recent findings: gut alpha-synuclein, microbiome changes in Parkinson’s disease
Ehraz Anis, Aoji Xie, Lena Brundin, Patrik Brundin
Trends in Endocrinology & Metabolism.2022; 33(2): 147. CrossRef - Animal models of brain-first and body-first Parkinson's disease
Nathalie Van Den Berge, Ayse Ulusoy
Neurobiology of Disease.2022; 163: 105599. CrossRef - Microbiome Changes in Humans with Parkinson’s Disease after Photobiomodulation Therapy: A Retrospective Study
Brian Bicknell, Ann Liebert, Craig S. McLachlan, Hosen Kiat
Journal of Personalized Medicine.2022; 12(1): 49. CrossRef - Passive Immunization in Alpha-Synuclein Preclinical Animal Models
Jonas Folke, Nelson Ferreira, Tomasz Brudek, Per Borghammer, Nathalie Van Den Berge
Biomolecules.2022; 12(2): 168. CrossRef - Dietary Succinoglycan Riclin Improves Glycemia Control in Mice with Type 2 Diabetes
Zhao Ding, Yang Zhao, Junhao Liu, Wenhao Ge, Xi Xu, Shiming Wang, Jianfa Zhang
Journal of Agricultural and Food Chemistry.2022; 70(6): 1819. CrossRef - Role of Microbiota-Gut-Brain Axis in Regulating Dopaminergic Signaling
Sevag Hamamah, Armin Aghazarian, Anthony Nazaryan, Andras Hajnal, Mihai Covasa
Biomedicines.2022; 10(2): 436. CrossRef - Distribution of α-Synuclein Aggregation in the Peripheral Tissues
Yan-yan Li, Tian-tian Zhou, Yi Zhang, Nai-Hong Chen, Yu-He Yuan
Neurochemical Research.2022; 47(12): 3627. CrossRef - Effects of Subdiaphragmatic Vagotomy in the MPTP-induced Neurotoxicity in the Striatum and Colon of Mice
Jiajing Shan, Youge Qu, Jiancheng Zhang, Li Ma, Kenji Hashimoto
Clinical Psychopharmacology and Neuroscience.2022; 20(2): 389. CrossRef - Alpha-Synuclein Strain Variability in Body-First and Brain-First Synucleinopathies
Mie Kristine Just, Hjalte Gram, Vasileios Theologidis, Poul Henning Jensen, K. Peter R. Nilsson, Mikael Lindgren, Karoline Knudsen, Per Borghammer, Nathalie Van Den Berge
Frontiers in Aging Neuroscience.2022;[Epub] CrossRef - Urolithins: potential biomarkers of gut dysbiosis and disease stage in Parkinson's patients
María Romo-Vaquero, Emiliano Fernández-Villalba, Ana-Luisa Gil-Martinez, Lorena Cuenca-Bermejo, Juan Carlos Espín, María Trinidad Herrero, María Victoria Selma
Food & Function.2022; 13(11): 6306. CrossRef - Short chain fatty acids-producing and mucin-degrading intestinal bacteria predict the progression of early Parkinson’s disease
Hiroshi Nishiwaki, Mikako Ito, Tomonari Hamaguchi, Tetsuya Maeda, Kenichi Kashihara, Yoshio Tsuboi, Jun Ueyama, Takumi Yoshida, Hiroyuki Hanada, Ichiro Takeuchi, Masahisa Katsuno, Masaaki Hirayama, Kinji Ohno
npj Parkinson's Disease.2022;[Epub] CrossRef - Opicapone, a Novel Catechol-O-methyl Transferase Inhibitor, for Treatment of Parkinson’s Disease “Off” Episodes
Amnon A. Berger, Ariel Winnick, Jonathan Izygon, Binil M. Jacob, Jessica S. Kaye, Rachel J. Kaye, Elisa E. Neuchat, Adam M. Kaye, Edward S. Alpaugh, Elyse M. Cornett, Andrew H. Han, Alan D. Kaye
Health Psychology Research.2022;[Epub] CrossRef - The role of LRRK2 in the periphery: link with Parkinson's disease and inflammatory diseases
George Tsafaras, Veerle Baekelandt
Neurobiology of Disease.2022; 172: 105806. CrossRef - What Is the Prognostic Significance of Rapid Eye Movement Sleep Without Atonia in a Polysomnogram?
Frank Ralls, Lisa Cutchen, Madeleine M. Grigg-Damberger
Journal of Clinical Neurophysiology.2022; 39(5): 346. CrossRef - Lippia grata essential oil complexed with β-cyclodextrin ameliorates biochemical and behavioral deficits in an animal model of progressive parkinsonism
Jose Ivo A. Beserra-Filho, Amanda Maria-Macêdo, Suellen Silva-Martins, Ana Cláudia Custódio-Silva, Beatriz Soares-Silva, Sara Pereira Silva, Rafael Herling Lambertucci, Adriano Antunes de Souza Araújo, Angélica Maria Lucchese, Lucindo J. Quintans-Júnior,
Metabolic Brain Disease.2022; 37(7): 2331. CrossRef - Oral and gut dysbiosis leads to functional alterations in Parkinson’s disease
Sungyang Jo, Woorim Kang, Yun Su Hwang, Seung Hyun Lee, Kye Won Park, Mi Sun Kim, Hyunna Lee, Hyung Jeong Yoon, Yoo Kyoung Park, Mauricio Chalita, Je Hee Lee, Hojun Sung, Jae-Yun Lee, Jin-Woo Bae, Sun Ju Chung
npj Parkinson's Disease.2022;[Epub] CrossRef - Deciphering Intertwined Molecular Pathways Underlying Metabolic Syndrome Leading to Parkinson’s Disease
Ritu Soni, Jigna Shah
ACS Chemical Neuroscience.2022; 13(15): 2240. CrossRef - Therapeutic potential of short-chain fatty acid production by gut microbiota in neurodegenerative disorders
Sarika Yadav, Ashish Dwivedi, Anurag Tripathi, Amit Kumar Tripathi
Nutrition Research.2022; 106: 72. CrossRef - Development of neurodegenerative disorders and the role of gut
Woong-Woo Lee
Journal of Geriatric Neurology.2022; 1(2): 53. CrossRef - What Are the Key Gut Microbiota Involved in Neurological Diseases? A Systematic Review
Bruno Bonnechère, Najaf Amin, Cornelia van Duijn
International Journal of Molecular Sciences.2022; 23(22): 13665. CrossRef - Gut microbiota in dementia with Lewy bodies
Hiroshi Nishiwaki, Jun Ueyama, Kenichi Kashihara, Mikako Ito, Tomonari Hamaguchi, Tetsuya Maeda, Yoshio Tsuboi, Masahisa Katsuno, Masaaki Hirayama, Kinji Ohno
npj Parkinson's Disease.2022;[Epub] CrossRef - Gut dysbiosis in stroke and its implications on Alzheimer’s disease‐like cognitive dysfunction
Justin Cho, You Jeong Park, Bella Gonzales‐Portillo, Madeline Saft, Blaise Cozene, Nadia Sadanandan, Cesar V. Borlongan
CNS Neuroscience & Therapeutics.2021; 27(5): 505. CrossRef - Plasma Lipopolysaccharide-Binding Protein Reflects Risk and Progression of Parkinson’s Disease
Szu-Ju Chen, Yu-Chiao Chi, Chang-Han Ho, Wei-Shiung Yang, Chin-Hsien Lin
Journal of Parkinson's Disease.2021; 11(3): 1129. CrossRef - Parkinson's disease and the gut: Models of an emerging relationship
Adam J. Bindas, Subhash Kulkarni, Ryan A. Koppes, Abigail N. Koppes
Acta Biomaterialia.2021; 132: 325. CrossRef - Ageing promotes pathological alpha-synuclein propagation and autonomic dysfunction in wild-type rats
Nathalie Van Den Berge, Nelson Ferreira, Trine Werenberg Mikkelsen, Aage Kristian Olsen Alstrup, Gültekin Tamgüney, Páll Karlsson, Astrid Juhl Terkelsen, Jens Randel Nyengaard, Poul Henning Jensen, Per Borghammer
Brain.2021; 144(6): 1853. CrossRef - Desulfovibrio Bacteria Are Associated With Parkinson’s Disease
Kari E. Murros, Vy A. Huynh, Timo M. Takala, Per E. J. Saris
Frontiers in Cellular and Infection Microbiology.2021;[Epub] CrossRef - “Janus-Faced” α-Synuclein: Role in Parkinson’s Disease
Bipul Ray, Arehally M. Mahalakshmi, Sunanda Tuladhar, Abid Bhat, Asha Srinivasan, Christophe Pellegrino, Anbarasu Kannan, Srinivasa Rao Bolla, Saravana Babu Chidambaram, Meena Kishore Sakharkar
Frontiers in Cell and Developmental Biology.2021;[Epub] CrossRef - Influence of probiotic bacteria on gut microbiota composition and gut wall function in an in-vitro model in patients with Parkinson's disease
Jonas Ghyselinck, Lynn Verstrepen, Frédéric Moens, Pieter Van Den Abbeele, Arnout Bruggeman, Jawal Said, Barry Smith, Lynne Ann Barker, Caroline Jordan, Valentina Leta, K. Ray Chaudhuri, Abdul W. Basit, Simon Gaisford
International Journal of Pharmaceutics: X.2021; 3: 100087. CrossRef - The Role of The Gut Microbiome in Parkinson’s Disease
Amy Gallop, James Weagley, Saif-ur-Rahman Paracha, George Grossberg
Journal of Geriatric Psychiatry and Neurology.2021; 34(4): 253. CrossRef - The Baseline Structure of the Enteric Nervous System and Its Role in Parkinson’s Disease
Gianfranco Natale, Larisa Ryskalin, Gabriele Morucci, Gloria Lazzeri, Alessandro Frati, Francesco Fornai
Life.2021; 11(8): 732. CrossRef - Regulation of neurotoxicity in the striatum and colon of MPTP-induced Parkinson’s disease mice by gut microbiome
Jiajing Shan, Youge Qu, Siming Wang, Yan Wei, Lijia Chang, Li Ma, Kenji Hashimoto
Brain Research Bulletin.2021; 177: 103. CrossRef - Cholecystectomy and subsequent risk of Parkinson’s disease: a nationwide retrospective cohort study
Ryul Kim, Jee-Young Lee, Sanghyun Park, Kyungdo Han, Cheol Min Shin
npj Parkinson's Disease.2021;[Epub] CrossRef - Benefits of Helicobacter pylori Eradication on Extragastric Diseases
Tae Jun Kim, Hyuk Lee
The Korean Journal of Helicobacter and Upper Gastrointestinal Research.2021; 21(4): 275. CrossRef - Higher cerebrospinal fluid to plasma ratio of p-cresol sulfate and indoxyl sulfate in patients with Parkinson’s disease
Bartłomiej Sankowski, Karolina Księżarczyk, Emilia Raćkowska, Stanisław Szlufik, Dariusz Koziorowski, Joanna Giebułtowicz
Clinica Chimica Acta.2020; 501: 165. CrossRef - The microbiome-gut-brain axis in acute and chronic brain diseases
Corinne Benakis, Camille Martin-Gallausiaux, Jean-Pierre Trezzi, Philip Melton, Arthur Liesz, Paul Wilmes
Current Opinion in Neurobiology.2020; 61: 1. CrossRef - The progress of gut microbiome research related to brain disorders
Sibo Zhu, Yanfeng Jiang, Kelin Xu, Mei Cui, Weimin Ye, Genming Zhao, Li Jin, Xingdong Chen
Journal of Neuroinflammation.2020;[Epub] CrossRef - Minireview on the Relations between Gut Microflora and Parkinson’s Disease: Further Biochemical (Oxidative Stress), Inflammatory, and Neurological Particularities
Ovidiu-Dumitru Ilie, Alin Ciobica, Jack McKenna, Bogdan Doroftei, Ioannis Mavroudis, Tuane B. Sampaio
Oxidative Medicine and Cellular Longevity.2020; 2020: 1. CrossRef - Microbiota-gut-brain axis in health and disease: Is NLRP3 inflammasome at the crossroads of microbiota-gut-brain communications?
Carolina Pellegrini, Luca Antonioli, Vincenzo Calderone, Rocchina Colucci, Matteo Fornai, Corrado Blandizzi
Progress in Neurobiology.2020; 191: 101806. CrossRef - Meta‐Analysis of Gut Dysbiosis in Parkinson's Disease
Hiroshi Nishiwaki, Mikako Ito, Tomohiro Ishida, Tomonari Hamaguchi, Tetsuya Maeda, Kenichi Kashihara, Yoshio Tsuboi, Jun Ueyama, Teppei Shimamura, Hiroshi Mori, Ken Kurokawa, Masahisa Katsuno, Masaaki Hirayama, Kinji Ohno
Movement Disorders.2020; 35(9): 1626. CrossRef - In Search of Effective Treatments Targeting α-Synuclein Toxicity in Synucleinopathies: Pros and Cons
Maria Fouka, Panagiota Mavroeidi, Grigoria Tsaka, Maria Xilouri
Frontiers in Cell and Developmental Biology.2020;[Epub] CrossRef - Role of Carbon Monoxide in Host–Gut Microbiome Communication
Christopher P. Hopper, Ladie Kimberly De La Cruz, Kristin V. Lyles, Lauren K. Wareham, Jack A. Gilbert, Zehava Eichenbaum, Marcin Magierowski, Robert K. Poole, Jakob Wollborn, Binghe Wang
Chemical Reviews.2020; 120(24): 13273. CrossRef - Neurodegeneration and Inflammation—An Interesting Interplay in Parkinson’s Disease
Chrysoula Marogianni, Maria Sokratous, Efthimios Dardiotis, Georgios M. Hadjigeorgiou, Dimitrios Bogdanos, Georgia Xiromerisiou
International Journal of Molecular Sciences.2020; 21(22): 8421. CrossRef - Niacin and Butyrate: Nutraceuticals Targeting Dysbiosis and Intestinal Permeability in Parkinson’s Disease
Tennekoon B. Karunaratne, Chijioke Okereke, Marissa Seamon, Sharad Purohit, Chandramohan Wakade, Amol Sharma
Nutrients.2020; 13(1): 28. CrossRef - Short-Chain Fatty Acid-Producing Gut Microbiota Is Decreased in Parkinson’s Disease but Not in Rapid-Eye-Movement Sleep Behavior Disorder
Hiroshi Nishiwaki, Tomonari Hamaguchi, Mikako Ito, Tomohiro Ishida, Tetsuya Maeda, Kenichi Kashihara, Yoshio Tsuboi, Jun Ueyama, Teppei Shimamura, Hiroshi Mori, Ken Kurokawa, Masahisa Katsuno, Masaaki Hirayama, Kinji Ohno, Chaysavanh Manichanh
mSystems.2020;[Epub] CrossRef - The Intestinal Barrier in Parkinson’s Disease: Current State of Knowledge
Sven C. D. van IJzendoorn, Pascal Derkinderen, Teus van Laar
Journal of Parkinson's Disease.2019; 9(s2): S323. CrossRef - Medical management, prevention and mitigation of environmental risks factors in Neurology
J. Reis, G.C. Román, M. Giroud, V.S. Palmer, P.S. Spencer
Revue Neurologique.2019; 175(10): 698. CrossRef - Antibiotic-induced microbiome depletion protects against MPTP-induced dopaminergic neurotoxicity in the brain
Yaoyu Pu, Lijia Chang, Youge Qu, Siming Wang, Kai Zhang, Kenji Hashimoto
Aging.2019; 11(17): 6915. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite- Figure
-

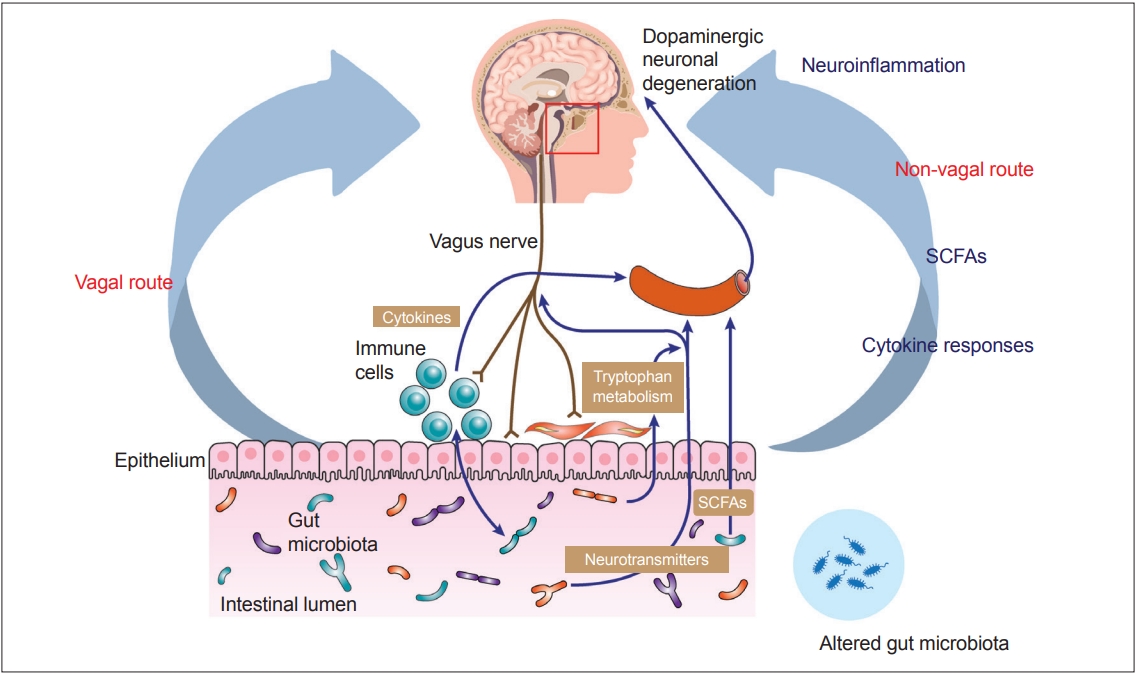
- Related articles
-
- Association of Depression With Early Occurrence of Postural Instability in Parkinson’s Disease
- Potential Link Between Cognition and Motor Reserve in Patients With Parkinson’s Disease
- Hand Movement-Induced Eyeblink Bursts in a Patient With Parkinson’s Disease
- Umami and Other Taste Perceptions in Patients With Parkinson’s Disease
- The Supplementary Motor Complex in Parkinson’s Disease
