E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 14(1); 2021 > Article
-
Review Article
Immune-Mediated Cerebellar Ataxias: Clinical Diagnosis and Treatment Based on Immunological and Physiological Mechanisms -
Hiroshi Mitoma1
 , Mario Manto2,3, Marios Hadjivassiliou4
, Mario Manto2,3, Marios Hadjivassiliou4 -
Journal of Movement Disorders 2021;14(1):10-28.
DOI: https://doi.org/10.14802/jmd.20040
Published online: January 12, 2021
1Department of Medical Education, Tokyo Medical University, Tokyo, Japan
2Service de Neurologie, Médiathèque Jean Jacquy, CHU-Charleroi, Charleroi, Belgium
3Service des Neurosciences, University of Mons, Mons, Belgium
4Academic Department of Neurosciences, Royal Hallamshire Hospital, Sheffield, UK
- Corresponding author: Hiroshi Mitoma, MD, PhD Department of Medical Education, Tokyo Medical University, Tokyo, Japan / Tel: +81-3-3345-5319 / Fax: +81-3-5339-3785 / E-mail: mitoma@tokyomed.ac.jp
Copyright © 2021 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Since the first description of immune-mediated cerebellar ataxias (IMCAs) by Charcot in 1868, several milestones have been reached in our understanding of this group of neurological disorders. IMCAs have diverse etiologies, such as gluten ataxia, postinfectious cerebellitis, paraneoplastic cerebellar degeneration, opsoclonus myoclonus syndrome, anti-GAD ataxia, and primary autoimmune cerebellar ataxia. The cerebellum, a vulnerable autoimmune target of the nervous system, has remarkable capacities (collectively known as the cerebellar reserve, closely linked to plasticity) to compensate and restore function following various pathological insults. Therefore, good prognosis is expected when immune-mediated therapeutic interventions are delivered during early stages when the cerebellar reserve can be preserved. However, some types of IMCAs show poor responses to immunotherapies, even if such therapies are introduced at an early stage. Thus, further research is needed to enhance our understanding of the autoimmune mechanisms underlying IMCAs, as such research could potentially lead to the development of more effective immunotherapies. We underscore the need to pursue the identification of robust biomarkers.
- History of immune ataxias: milestones
- Immune-mediated pathophysiological mechanisms frequently affect the cerebellum, leading to progressive ataxia characterized by dysmetria in both motor and cognitive domains [1,2]. The first documentation of patients with immune-mediated cerebellar ataxias (IMCAs) originated from Charcot [3]. In a well-documented lecture on multiple sclerosis (MS) delivered in 1868, he described the presence of cerebellar ataxia (CA) in patients with MS, now known as the Charcot’s triad (intention tremor, scanning speech, and nystagmus). Another historical milestone was the first report of paraneoplastic cerebellar degeneration (PCD) by Brouwer [4] in 1919, in which he described the association of CA with ovarian cancer.
- In the 1980s, the identification of autoantibodies targeting cerebellar neurons led to a breakthrough in IMCAs. First, the discovery of anti-Yo antibody (Ab), an autoantibody described in a patient with ovarian cancer, demonstrated the immune nature of the insult resulting in PCD [5]. This discovery was followed by the identification of specific autoantibodies, including anti-Hu, anti-Tr, anti-CV2, anti-Ri, anti-Ma2, and anti-VGCC Abs, often associated with specific types of neoplasms, including breast, uterine, ovarian, small cell lung carcinoma, and Hodgkin’s lymphoma [6]. Second, the association of otherwise idiopathic CAs with autoantibodies against the cerebellum was also reported in patients without evidence of cancer [7-11]. Two main clinical entities have been established since then, based on specific clinical features and types of associated autoantibodies: gluten ataxia (GA) by Hadjivassiliou et al. [12] and anti-glutamic acid decarboxylase 65 Abs (GAD 65 Abs)-associated cerebellar ataxia (antiGAD ataxia) by Honnorat et al. [13].
- Finally, in 2008, Hadjivassiliou et al. [14] proposed a new clinical entity: primary autoimmune cerebellar ataxia (PACA). This entity encompasses all ataxic patients who exhibit features of IMCAs but with a serological profile that does not match any of the known main etiologies. Diagnostic criteria have been proposed recently [15]. Thus, it has become easier to apply this diagnosis for such atypical patients to consider the use of immunotherapy.
- Classification
- IMCAs gather diverse etiologies, and the pathophysiology is not restricted to the cerebellum, also targeting cerebellar-related structures/connections in the central nervous system (CNS). The diversity renders the classification of IMCAs challenging. We previously proposed a classification based on 2 criteria: 1) whether the cerebellum is the main target of autoimmunity, and 2) whether autoimmunity is generated by a known trigger or not (Table 1) [8-11].
- In MS, for example, the cerebellum is one of many targets within the CNS. Pure CA types, in which the cerebellum or its related structures are the sole target of autoimmunity, include GA, postinfectious cerebellitis (PIC), PCD, opsoclonus myoclonus syndrome (OMS), anti-GAD ataxia, and PACA. Thus, the term IMCAs is reserved for mainly pure or primarily CA, thus excluding more widespread autoimmune damage such as that seen in the context of MS. Although autoimmune limbic encephalitis can sometimes be characterized by CA in addition to cognitive/behavioral deficits and seizures, these conditions should not be categorized as IMCAs.
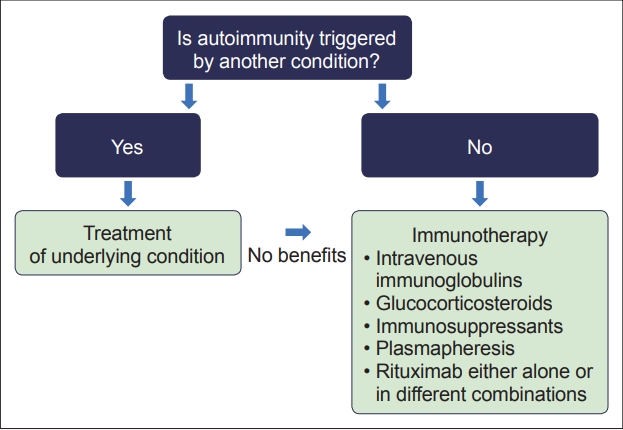
- Pure or primarily CA etiologies are divided into two groups: 1) etiologies in which autoimmunity is triggered by other conditions, such as infection (e.g., PIC), neoplasm (e.g., PCDs), and gluten sensitivity (GA), and 2) etiologies in which autoimmunity is not triggered by any other condition (e.g., anti-GAD ataxia). When autoimmunity is triggered by other conditions, removal of the trigger should be the first line of therapy. This approach is important because categorization based on the autoimmune triggering factors can also offer therapeutic strategies [9-11].
- In the case of PACA, immune-mediated mechanisms are strongly suspected, but the serological profile of the patients does not match any of the known diseases in IMCAs [14,15]. Thus, this group is likely to be heterogeneous and probably includes several pathophysiological immune-mediated mechanisms.
- Prognosis and pathophysiology
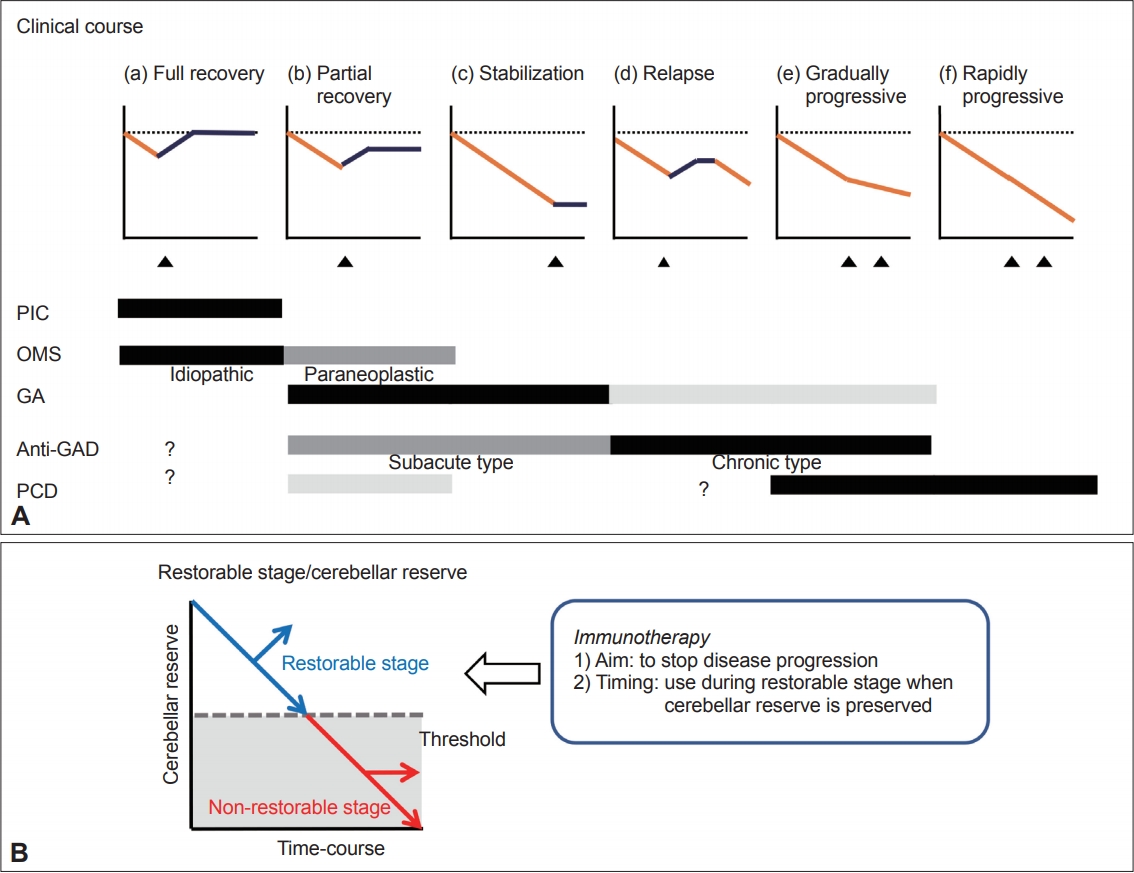
- In keeping with the diverse etiologies, IMCAs show characteristically diverse clinical courses (Figure 1) [10,11]. These diverse clinical courses result in two problems when considering therapeutic strategies: the need for early intervention and the unclear immune-mediated pathophysiology.
- GA, PIC and the subacute type of anti-GAD ataxia show good response to immunotherapy (full or partial recovery and stabilization) (Figure 1). The good prognosis is in part due to the benign transient autoimmune insult, which is also seen in other autoimmune diseases. Alternatively, the reversibility can also be attributed to specific cerebellar capacity for compensation and restoration following the immune insult [16,17]. We termed this capacity the cerebellar reserve [16,17]. Various forms of synaptic plasticity and redundant mossy fiber-mediated inputs constitute the cerebellar reserve [16,17]. Notably, the extent of progression of the pathophysiological mechanisms determines the degree of reversibility, suggesting the existence of a threshold. Although various immunotherapies can arrest the progression of the damage under this threshold, such intervention may not be accompanied by clinical improvement. Thus, immunotherapies should be introduced during the period when the cerebellar reserve is above this threshold. Such a threshold can be understood as the limit required to obtain a sufficient level of activity in the cerebellar circuitry.
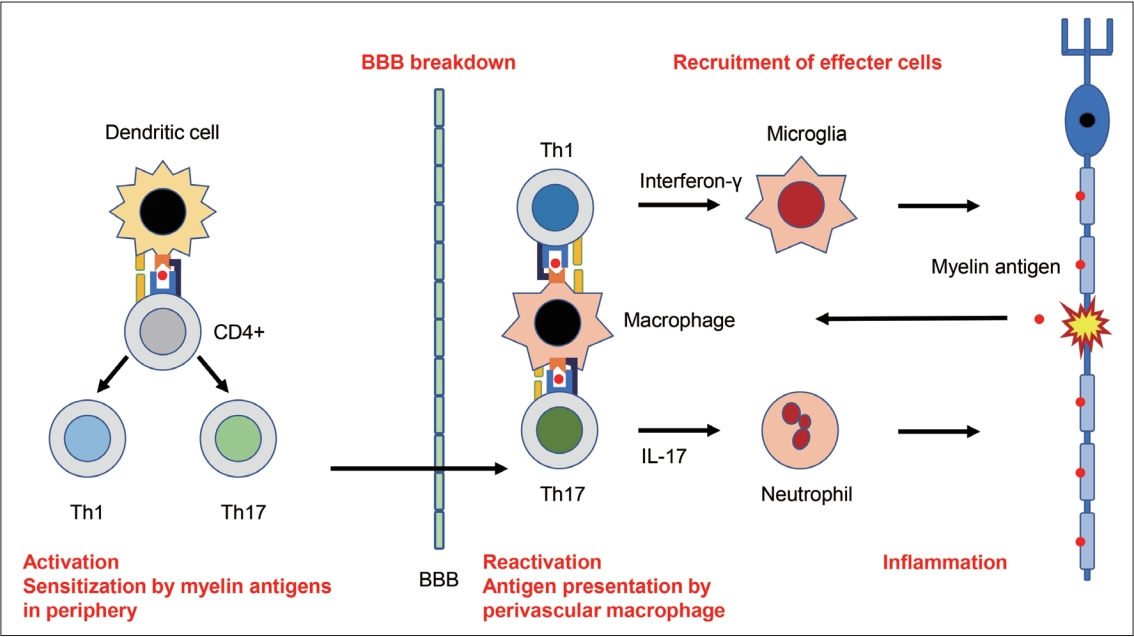
- The chronic subtypes of anti-GAD ataxia and PCD show poor prognosis (relapse and gradual or rapid progression in Figure 1), which reflects poor responsiveness to immunotherapy. The development of various autoimmune diseases can be attributed to common elementary processes [18], including autoimmunity triggered by deficits in immune tolerance or molecular mimicry, increased permeability of the blood-brain barrier or blood-nerve barrier, pathogenic actions of T cells (Th1/17 cells and CD8 T cells) or autoantibodies, and exacerbation of local neural inflammation (Figure 2). However, unlike the advances in the field of MS, little is known about therapeutic autoimmune targets in IMCAs.
- In conclusion, the clinical entity of IMCAs has been established during the past three decades. To avoid missing therapeutic opportunities at the early stage, we should carefully promote IMCAs in the differential diagnosis of all ataxias. We also need more research directed toward the identification of new autoimmune pathogenesis-based therapies.
BACKGROUND: WHERE DO WE STAND?
Cerebellar reserve and early intervention
Autoimmune mechanisms and response to treatment
- Prevalence
- Hadjivassiliou et al. [19] (2017) investigated the prevalence of IMCAs on 1,500 UK patients with progressive ataxia. The authors reported that 30% of the patients had familial/genetic ataxia, although some did not show evident family history, and 9% of the patients had a cerebellar variant of multiple systemic atrophy (prevalence out of total progressive ataxic cases). Apart from the above, 25% had definite IMCAs; 20% had GA, and 2% had PCD, while 2% had anti-GAD ataxia, 1% had PIC, and <1% had OMS. Interestingly, 19% of the patients were classified as idiopathic sporadic ataxia. This category probably included a large number of patients with PACA [14,15].
- Among other groups, Gebus et al. [20] (2017) reported a smaller number of IMCAs patients (2 with PCD and 1 patient with PIC among 80 patients). In a systematic study of 684 South Korean patients with progressive ataxia, only 21 had IMCAs; 14 patients with PCD, 3 patients with PIC, and 4 patients other immune causes [21]. The discrepancy might be attributed to a number of different factors: The high prevalence of IMCAs in Hadjivassiliou’s study [19] was due to a large number of patients with GA. GA was first characterized and reported by this group and there is therefore the potential for referral bias. Another important factor is the serological test utilized to make a diagnosis of GA (please see section on GA). More recently, the prevalence of GA in Asian countries has been found to be higher than was previously estimated [22,23]. For example, patients with GA have recently been reported in Japan [23]. A survey in China also showed that the prevalence rates of positive-anti-transglutaminase 6 (TG6) IgA were 35% in 100 sporadic CA patients and 9.8% in 51 healthy controls (p = 0.001) [23]. Another factor is the so called “idiopathic” unknown etiology diagnosis. As discussed above, we have argued that this group includes a large number of patients with PACA [8-11]. The newly established diagnostic criteria on PACA [15] will be helpful in resolving this issue.
- Further studies are needed to examine the prevalence of IMCAs or GA in progressive CAs. However, early diagnosis and intervention is critical during a period when the cerebellar reserve is preserved. In this regard, clinicians should always consider the possibility of IMCAs in the differential diagnosis of patients developing CAs.
- Clue to diagnosis
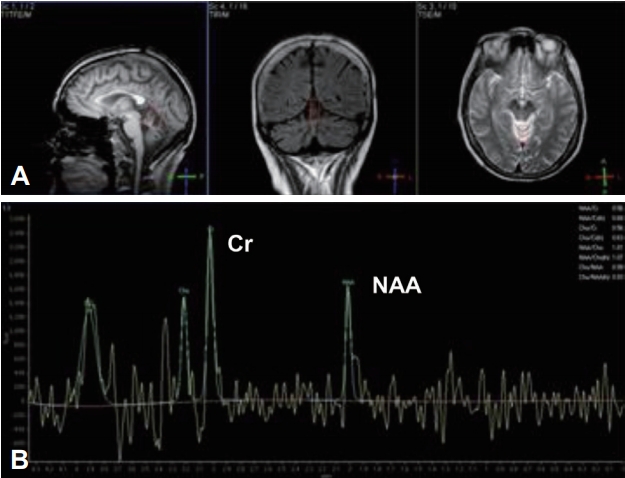
- Patients with IMCAs exhibit common clinical manifestations in spite of divergent etiology [10,11]. The onset of CA is usually acute/subacute or sometimes chronic/insidious. The presence of other autoimmune disorders is common in some patients. The main clinical feature is gait ataxia, which influences standing and steady walking and is associated with mild limb incoordination and dysmetria, oculomotor ataxia, and scanning speech. In keeping with these symptoms, MRI characteristically shows vermian atrophy, which can be a clue for the differential diagnosis from degenerative or genetic CAs (Figure 3A). However, it should be acknowledged that MRI can be normal. As the pathology progresses, atrophy becomes more evident, preferentially affecting the vermis. CSF studies sometimes show the presence of pleocytosis or oligoclonal bands. There may be a context of systemic impaired immunity (affecting skin, joints or other organs).
- Autoantibodies
- As discussed earlier in the section on the history of IMCAs, identification of autoantibodies is important in the diagnosis (Table 2) [10,11]. Immunohistochemistry may reveal the binding of autoantibodies toward cerebellar components; therefore, if available, it can add further evidence of the diagnosis. Autoantibodies in IMCAs can be divided into two categories: 1) autoantibodies suggestive of specific etiologies, and 2) nonspecific autoantibodies found in other neurological and systemic conditions, including CAs, which provide only possible autoimmune pathophysiology. The former types of autoantibodies include antigliadin and TG6 Abs in GA, and anti-Yo, Hu, CV2, Ri, and Ma2 Abs for PCD [5,6,8,10,11,24]. The nonspecific autoantibodies are also subdivided into two subcategories: autoantibodies that are assumed to have pathogenic roles and the nonpathogenic autoantibodies (i.e., a diagnostic marker).
- The majority of the pathogenic autoantibodies include autoantibodies toward ion channels or ion channel-related proteins (Ca++ or K+ channel), glutamate receptors, or GABA synthesis enzyme (GAD), some of which have been confirmed as pathogenic in both in vitro and in vivo preparations [24-34]. Impairments of neural excitability and synaptic transmissions can occur throughout the nervous system, such as limbic encephalitis [24-34]. We previously proposed a nomenclature based on the pathogenic role of the autoantibody, i.e., the pathogenic Ab to be coined to the name of the clinical entity (e.g., anti-GAD ataxia) [10,11].
- Additionally, some autoantibodies are reported to be associated with CAs in only a few patients. These autoantibodies are classified into a category of not-well-characterized autoantibodies. The significance of these autoantibodies remains to be determined, but their presence can be a clue to the autoimmune etiology of the ataxia.
- Principles of treatment
- When autoimmunity is caused by another trigger, priority should be given to the treatment of the underlying condition: for example, gluten-free diet (GFD) in GA and surgical excision or chemotherapy and radiotherapy of the neoplasm in PCD (Figure 4) [7-11]. For other IMCAs, immediate immunotherapy is recommended (Figure 4) [7-11].
- Depending on the rapidity of progression, induction immunotherapy can be used to arrest progression of CA, followed by maintenance immunotherapy to prevent relapse [9-11]. Various immunotherapies have been used, ranging from intravenous immunoglobulins (IVIg), glucocorticosteroids, mycophenolate, plasmapheresis, and rituximab, either alone or in combination, and the selection is often based on the etiology and presentation [9-11]. These therapies should be introduced during the period when the cerebellar reserve is still preserved.
- To establish a therapeutic rationale, the following two issues need further consideration. First, to date, there are no large-scale randomized studies comparing therapeutic strategies for each IMCA [9-11]. Second, the effects of each therapeutic intervention should be quantified with more sensitive biomarkers if possible at an early stage of the disease when MRI shows no atrophy. Recent studies highlighted the potential usefulness of MR spectroscopy as a sensitive biological marker of the response to treatment in CA patients [10,11]. The ratio of N-acetylaspartate (NAA)/creatine (Cr) area is decreased in patients with CAs (Figure 3B) relative to the control and increases in those who respond to immunotherapy and exhibit clinical improvement [10,11].
GENERAL ISSUES ON DIAGNOSIS AND IMMUNOTHERAPIES
- This section focuses on main etiologies in IMCAs (Table 3).
- Gluten ataxia
- GA is defined as sporadic CA associated with gluten sensitivity [12]. The autoimmunity is triggered by the sensitivity to gluten found in wheat, rye and barley. Gluten, a family of protein containing grains, is composed of gliadin and glutenin.
- GA affects individuals in their 40–50s and exhibits either chronic or insidious onset [7-11]. Gluten-sensitive enteropathy (coeliac disease) is seen in approximately half of the patients [7-11]. The human leukocyte antigen (HLA) type DQ2 is detected in 70% of the patients, and association with other autoimmune diseases, such as thyroiditis, type 1 diabetes mellitus, and pernicious anemia, is common [7-11]. Patients present with gait ataxia and variable degree of limb ataxia, scanning speech [7-11] and, consistently, MRI shows vermian involvement [35]. GA can be associated with CSF abnormalities, with oligoclonal bands in up to 50% of patients [7-11]. The association of sensorimotor axonal neuropathy and focal myoclonus, rarely palatal tremor, is sometimes observed [7-11].
- Diagnosis is based on the presence of serological evidence of gluten sensitivity, which is assessed using autoantibody testing. Native anti-gliadin Ab (AGA) remains to be the most reliable test for the diagnosis [10,11]. However, the cutoff level for the diagnosis of GA should be noted [36]. In GA patients without enteropathy, the main autoimmune response occurs within the CNS, not the periphery, resulting in low levels of serum AGA [10]. The calibration process used in certain commercially available AGA kits is based on the use of serum from patients with celiac disease (CD) and, therefore, the cutoff level is too high for the detection of GA [10]. Anti-transglutaminase 2 (TG2) Ab, found in patients with CD, is negative in 53% of GA patients free of enteropathy [36]. However, anti-TG6 Ab is positive in 72% of GA patients on the basis of AGA [37]. Since TG6 is primarily expressed in the CNS, anti-TG6 Ab could be an important and possibly more specific biomarker [10,11].
- GFD is the first line of therapy for GA since it can eliminate antigens that can trigger immune-mediated mechanisms, similar to the strategy used in CD [7-11]. One large-scale study based on 43 patients with GA showed significant improvements in CA and a decrease in AGA in patients who adhered to GFD, compared with those who did not [38]. Other reports described the effectiveness of IVIg in nonresponders to GFD [39]. However, it is now considered that the lack of response to GFD is due to either poor adherence to GFD or hypersensitivity to gluten, where a small amount of gluten present in commercially available gluten-free food or due to cross-contamination can cause strong autoimmune reactions perpetuating the cerebellar damage [9-11]. Persistently high levels of AGA are present in the above two groups of patients [40,41]. On MR spectroscopy, patients on strict GFD show an increase in the ratio of NAA/Cr area in the cerebellar vermis, whereas no such increase is seen in patients on GFD with persistently positive antibodies and those who do not adhere to GFD [42]. Taken together, in patients with persistently high AGA titers or no improvement in MR spectroscopy, further dietetic review by an expert dietitian should be considered before switching to immunotherapy [10,11]. When CAs cannot be controlled with GFD, maintenance therapy with IVIg or immunosuppressants (e.g., mycophenolate mofetil, cyclosporin, and cyclophosphamide) is recommended [9-11].
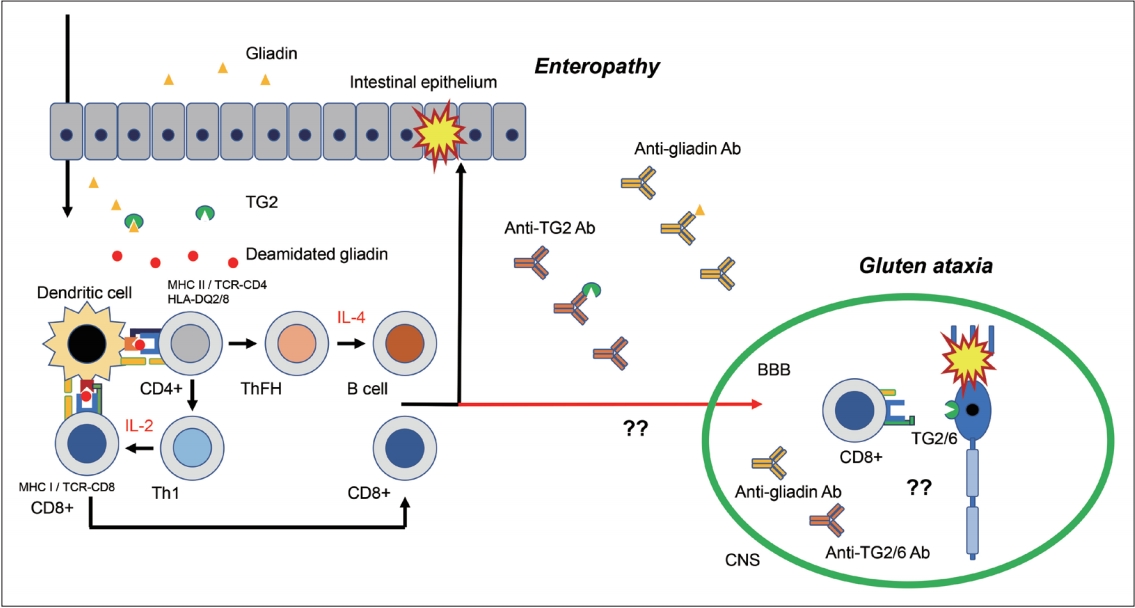
- Loss of immune tolerance is considered the main mechanism, as in coeliac disease [43]. Digested gliadin is deamidated and cross-linked by TG2, leading to the creation of an immunostimulatory epitope for HLA-DQ2 or HLA-DQ8 on antigen-presenting cells. These epitopes are presented to CD4+ T cells, from which cytokines are released to facilitate the production of antibodies against gliadin and TG2 (TG2-gliadin complex) (Figure 5).
- The neural damage appears to be due to, at least in part, the actions of autoantibodies against tissue transglutaminase, a calcium-dependent enzyme located in intra- and extracellular spaces. Both anti-TG2 and anti-TG6 Abs recognize and cross-react with neurons. Intraventricular administration of anti-TG2 or anti-TG6 Abs elicits ataxia in mice [44]. TG2 activity is upregulated through calcium binding, and anti-TG2 Abs bind preferentially to the calcium-activated enzyme conformation [45,46]. Taken together, antibody-induced deficits of TG2 in its enzymatically active conformation can cause cellular dysfunction. The role of TG6 in cerebellar function is also supported by the observation that mutations in the TGM6 gene can cause CA (spinocerebellar ataxia type 35) [47].
- Although the mechanisms of cell-mediated brain damage remain uncertain, the above immunological studies add support to the therapeutic rationale of gluten avoidance in food and reductions of anti-TG2/6 and AGAs.
- CA associated with cortical myoclonus is a rare subtype of gluten sensitivity-related neurological disorder [10,11,48]. Although the myoclonus is of cortical origin, hyperexcitability of the cerebral cortex is elicited by cerebellar dysfunction [10,11,48,49]. Characteristically, this subtype shows resistance to GFD. The neurological refractoriness is associated with residual enteropathy, which is detected by repeat duodenal biopsies [9-11]. All such patients require GFD plus immunosuppression, usually with mycophenolate and in some instances cladribine, in addition to anti-epileptic drugs for the control of the myoclonus [9-11]. The prognosis remains poor [10,11].
- Postinfectious cerebellitis
- PIC is defined as cerebellar inflammation induced by immune-mediated mechanisms triggered by viral or bacterial infection [7,8,10,11,50]. Some groups have termed this type of autoimmune ataxia as para-infectious cerebellitis or acute cerebellar ataxia [10,11,50,51]. However, when the cerebellar inflammation is caused by direct invasion of viral or bacterial microorganisms, it is called acute cerebellitis [10,11,52].
- PIC affects mostly young children after an episode of infection, usually viral infection, most commonly varicella [7,8,10,50-52]. Other viruses, such as Epstein-Barr virus, Coxsackie virus, influenza A and B virus, parainfluenza virus, rubeola virus (measles), rubulavirus (mumps), and rubella virus (German measles), and bacteria, such as Corynebacterium diphtheriae (diphtheria), Bordetella pertussis (whooping cough), Salmonella typhi (typhoid fever), Legionella (Legionnaires disease), Leptospira (leptospirosis), and Mycoplasma (mycoplasmosis) can be involved [7,8,10,50,52]. Borrelia (Lyme disease) is also implicated. One large-scale study based on 73 patients [53] showed that 60% of the patients were between 2 and 4 years of age, and 25% of these patients had varicella, 52% had other viral infections, and 3% developed PIC after immunization. The mean latency between infection and the onset of CAs was 9.9 ± 7.9 days. However, some patients (19%) showed no definite preceding infection.
- Patients develop mainly acute-onset afebrile gait ataxia, meningeal signs, high intracranial pressure with or without extracerebellar manifestations, such as temporary clouding of consciousness, seizures, altered mental status (e.g., extreme irritability), or extracerebellar focal signs [53]. The presence of these clinical features is often suggestive of direct infective etiology rather than immune-mediated mechanisms (PIC) [52]. Additionally, mild behavioral changes, such as mild irritability, hyperactivity, moodiness and whining, are sometimes noted by parents and often correlate with the severity of CA [52]. These manifestations could reflect cerebellar cognitive and emotional disorders. CSF studies show pleocytosis in most of the patients (roughly equal numbers of granulocytes and lymphocytes in approximately 75% of the patients, and sometimes lymphocyte predominance in the remaining cases) and high CSF/serum IgG index in half of the children. Oligoclonal bands are sometimes present [53]. MRI is usually negative, with no signs of atrophy or abnormal intensity areas [53]. Nevertheless, adult patients can develop cerebellar atrophy after several months.
- Since PIC is self-limiting, close observation of the patient is recommended [10,11]. One large-scale study reported full recovery of CAs in 72% of 60 pediatric patients within two months [53]. Only when the CAs persist or progress are various combinations of immunotherapeutic agents available for use [10,11,50].
- Autoimmune mechanisms remain obscure. For example, it is not certain whether molecular mimicry, observed in GuillainBarré syndrome (GBS, a typical infection-induced neuroimmune disease), also operates in PIC. Although the lack of cerebellar atrophy and self-limiting clinical course suggests immunemediated cerebellar functional disorders, the exact mechanisms remain unclear. A recent study [54] described the unique actions of anti-GluRδ Ab, which is associated with PIC in some patients [26]. In one experimental study, injection of an antibody toward the putative ligand-binding site of GluRδ2 resulted in endocytosis of AMPA receptors and attenuated synaptic transmission and ultimately led to the development of ataxic behavior in mice [54]. Interestingly, the endocytosis of AMPA receptors, which has a transient nature, could explain the reversibility in PIC. Thus, further studies are needed to understand how autoimmune reactions transiently disturb cerebellar function.
- Opsoclonus myoclonus syndrome
- OMS has divergent categories, including paraneoplastic, postinfectious, and idiopathic [10,11,55-57]. It affects mainly children and is especially associated with neuroblastoma [10,11,55-57].
- Opsoclonus is characterized by involuntary repetitive, random and rapid eye movements in both horizontal and vertical directions. Action myoclonus is characterized by irregular jerky movements primarily in upper limbs [10,11]. Since OMS is usually associated with CA, it is known also as opsoclonus myoclonus ataxia syndrome [10,11]. In keeping with the diverse etiology, some differences are observed in the clinical profiles of patients with OMS. The presence of subacute vertigo at onset, together with CSF abnormalities (high leukocyte count and protein content), is more common (half of the patients) in idiopathic OMS than in paraneoplastic OMS. Encephalopathy is more common in paraneoplastic OMS, although it is also found in at least a few cases of idiopathic OMS [55]. Anti-Ri Ab, an onconeural Ab, is positive in paraneoplastic patients, mainly those with breast cancer [55]. Onconeural Ab and PET scan should be performed to confirm any association with neoplasm [7,10,11].
- In cases of paraneoplastic OMS, any associated neoplasm should be removed first, if possible, followed by a combination of immunotherapies (corticosteroids, IVIg, plasmapheresis, immunosuppressants, and rituximab) [10,11,55-57]. Postinfectious OMS and some idiopathic OMS are self-limiting. Thus, if symptoms persist, immunotherapy should be introduced [10,11,55-57]. Several studies showed good prognosis in idiopathic OMS compared with paraneoplastic OMS [55-57]. For example, when a good response to immunotherapy was defined as recovery of the modified Rankin Score to ≤ 2 [55], a good response was reported in 39% of patients with paraneoplastic OMS and in 84% of those with idiopathic OMS. Relapse occurred in 24% of patients with paraneoplastic OMS and in 7% of those with idiopathic OMS [55].
- Autoimmune pathogenesis is based on the presence of CD19+ B cells and gamma-delta T cell subsets in the CSF [58] as well as perivascular collection of lymphocytes in autopsy cases [59]. It should be noted that unlike PCD, neither cell loss nor extensive T cell infiltration is characteristically evident in paraneoplastic OMS [60,61], which suggests immune-mediated functional disorders in the cerebellar motor controls.
- Characteristically in OMS, the autoimmune-induced dysfunction appears to occur not only within the cerebellum but also in inhibitory controls from the cerebellar cortex on cerebellar nuclei neurons. It is assumed that the net inhibitory output from Purkinje cells (PCs) in the cerebellar cortex is attenuated in both opsoclonus and action myoclonus. In ocular motor controls, this elicits hyperactivation of the fastigial nucleus neurons, which in turn facilitates the activities of excitatory burst neurons, leading to a chaotic cascade of events [62]. In the cerebro-cerebellar loop, the pathway of the dentato-thalamo-cortical tract is hyperactivated, in which even subthreshold inputs can easily activate the motor cortex, generating the action myoclonus in limb movements. Consistently, action myoclonus is of cortical origin [48,49], and a low correlation between the cerebellum and motor cortex is observed in functional MRI [63].
- In conclusion, there is no explanation on how the autoimmune processes, typically triggered by diverse pathologies, such as neoplasm and infection, commonly produce a unique functional deficit, decrease the inhibitory output from the cerebellar cortex and generate hyperexcitability of their targets.
- Paraneoplastic cerebellar degeneration
- PCD comprises cerebellar degeneration caused by neoplasmtriggered autoimmunity [5-8,24,64]. The presence of characteristic onconeural autoantibodies is not only diagnostic for PCD but also provides a clue to the specific type of neoplasm underlying PCD (Table 2). Although aggressive chemotherapies and immunotherapies have been attempted, the prognosis remains poor unless the underlying cancer can be completely and rapidly eradicated.
- PCD shows acute or subacute onset and is sometimes preceded by prodromal clinical symptoms such as nausea, vomiting, and dizziness, resembling viral infection-related disease [8,24,64]. Subsequently, patients show gait ataxia, which is followed by pancerebellar involvement [8,24,64]. CSF studies show changes consistent with inflammation, including moderate lymphocytic pleocytosis, high protein concentrations, high IgG index, and oligoclonal bands [8,24,64]. Identification of the characteristic autoantibodies helps in the establishment of diagnosis, although seronegative cases have been described [6]. Since subacute CAs are listed as classical symptoms defined in the 2004 diagnostic criteria [65], a definite diagnosis is based on 1) confirmation of the presence of cancer, which develops within 5 years of the diagnosis of CA, or 2) appearance of well-characterized onconeural Abs (anti-Yo, anti-Hu, anti-CV2, anti-Ri, anti-MA2) [65]. CA is the first manifestation of neoplasm in up to 70% of the patients with PCD [64], highlighting the need for systemic evaluation for malignant tumors following the identification of onconeuronal autoantibodies.
- Following confirmation of the diagnosis, anti-neoplasm therapies should be introduced immediately to prevent metastasis and remove antigen(s) driving the autoimmunity [8,24,64]. The selection of cancer treatment, such as surgery, radiotherapy and/or chemotherapy, is based on the type of the neoplasm [8,24,64]. Regardless of the selected cancer treatment modality, various types of immunotherapies (single or combinations of corticosteroids, IVIg, plasmapheresis, immunosuppressants, and rituximab) should be administered.
- In spite of these treatment protocols, the prognosis is poor [64,66-68]. The response to immunotherapy is generally unsatisfactory [64,66-68], although therapeutic benefits have been reported in some case reports [10], and better prognosis was noted in patients with anti-Tr Ab [69]. The study of Peterson et al. [70] on 22 patients with anti-Yo Ab reported therapeutic improvement in less than 10% of the trials (one in four trials of plasmapheresis, one in 17 trials of corticosteroids, and none in four trials of cyclophosphamide). Furthermore, Rojas et al. [71] also reported no benefits from the combinations of immunotherapies in their study of 23 patients with PCDs.
- There have been three long-term studies on survival time [69,72,73]. Candler et al. [72] examined the clinical courses of 63 patients with paraneoplastic neurological syndromes, including 13 patients with PCDs. Three of the 13 (23%) patients with PCDs died before the final follow-up, and the mean survival time from onset of neurological symptoms was 42 months (95% confidence interval: 32–52). Keime-Guibert et al. [73] reported a long-term follow-up study of 16 patients with PCDs. When the clinical benefits were determined as a more than one point change in the Rankin Score, none of their patients met this criteria (three patients showed stabilization). The median survival time from the first therapy was 10.2 months (range 2–38). Fourteen of the 16 (88%) patients died during the follow-up. The cause of death was cancer-related in 9 patients, neurological-related in 3 patients, and unknown in the remaining patients. Shams’ili et al. [69] examined the relationship between effectiveness of treatment and associated antibodies in their study of 50 patients with PCDs. The median survival times from diagnosis were 113 months in patients positive for anti-Tr Ab, > 69 months in those positive for anti-Ri Ab, 13 months in those positive for anti-Yo Ab, and 7 months in those positive for anti-Hu Ab. It should be noted that even patients with anti-Tr Ab suffered severe disability or dependence. Taken together, the survival time is generally short once the diagnosis is established, excluding a few patients with anti-Tr Ab.
- The poor prognosis could be attributed to the remnant tumor tissue and metastasis, which could cause persistent activation of the immune system and strong autoimmune response. Currently, clinical trials aimed at stopping the progressive autoimmune insults are being conducted. In one such trial, the combination of IVIg or plasmapheresis with cyclophosphamide was reported to be effective in a subgroup of patients [64].
- Pathological studies have identified various inflammatory changes in the early stages of PCD, including perivascular cuffing by lymphocytes, infiltration of lymphocytes within the PC layer, and microglial activation [74,75]. The infiltrates consist of both of T and B cells, plasma cells, microglia/macrophage lineages, and reactive Bergmann cell gliosis [76]. A similar infiltration, albeit of lower magnitude, was found in the dentate nucleus and the pons [77,78]. Disease progression is associated with massive loss of PC without inflammation (inflammatory cells or deposits of immunoglobulins and complements) [76,79,80]. Since the loss of PC is often very rapid, such a burnout stage can be observed in some patients at an earlier stage [76,79].
- Yo (Cdr2) and Hu onconeural antigens are intracellularly located in tumor and neuronal cells [81]. This favors molecular mimicry mechanisms. First, whether autoantibodies that target intracellular antigens Cdr and Hu proteins play pathogenic roles in the development of the inflammatory cerebellar lesions is still a matter of debate today. Previous studies showed that anti-Yo Ab toward Cdr2 was internalized and induced cell death through apoptosis [82,83]. In this regard, Cdr2 encodes a leucine zipper motif and interacts with another leucine zipper motif on c-Myc, the nuclear transcription factor, to regulate nuclear levels of c-Myc and c-Myc-associated signaling activities. Cdr2-specific anti-Yo Ab inhibits the interaction of Cdr2 with c-Myc, possibly allowing excess c-Myc to enter the nucleus and impair cell cycle signaling involved in cell apoptosis. The binding of anti-Yo Ab to Cdr2 can also disrupt interaction with the mortality factor-like proteins MRGX and/or NFkb, leading to changes in transcriptional activity [83]. Another study showed that administration of anti-Hu and Yo induced the expression of adhesion molecules and more intense cell differentiation rather than cell death [84]. Furthermore, another histochemical study showed a lack of both IgG deposits and B cell infiltration [85]. A study of passive transfer of onconeural Abs that targeted intracellular antigens and immunization using protein or DNA failed to induce ataxic symptoms in animals [86-88]. Thus, many researchers believe that antibody-mediated mechanisms are not the main autoimmune mechanisms underlying PCDs associated with anti-Yo and anti-Hu Abs [64,85].
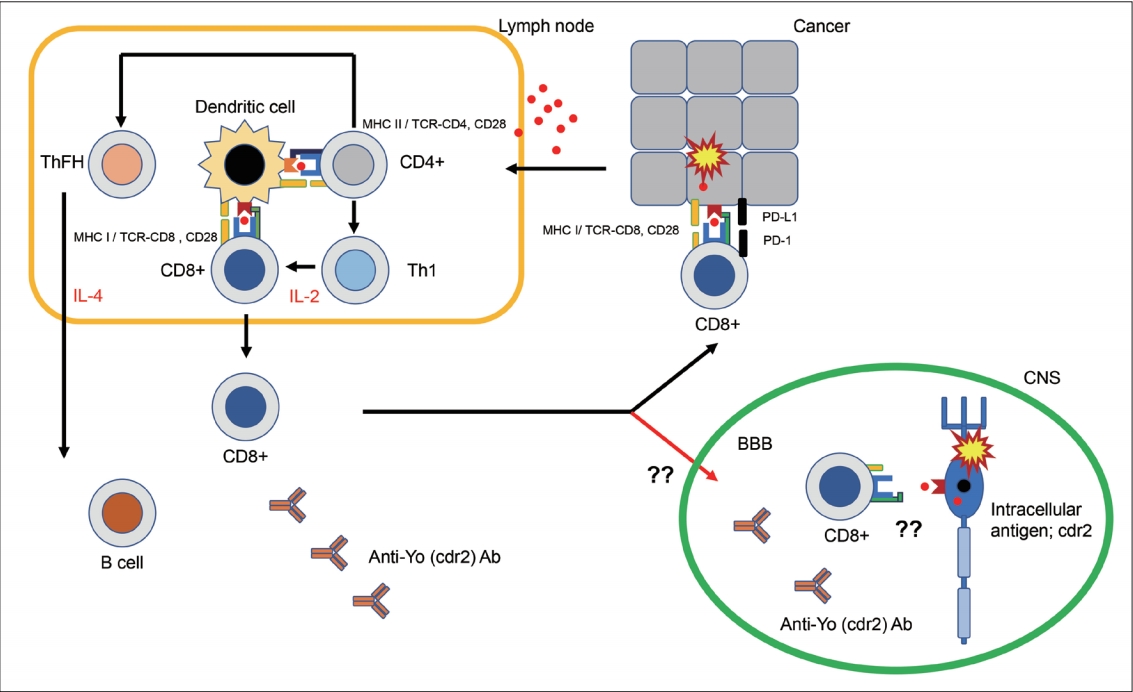
- Patients with anti-Yo (Cdr2) Ab or anti-Hu Ab show accumulation of Cdr2- or Hu-specific T cells in blood and CSF, respectively [89-91]. Notably, the lymphocytes found in tumors, peripheral blood, and affected nervous tissue are thought to originate from the same clone population [81,92]. The infiltrated lymphocytes in the cerebellum are mainly CD3+ and CD8+ T cells [78,92], and only a few are CD4+ lymphocytes [93]. In general, CD4+ and CD8+ T cell interaction is necessary to induce inflammation, where CD4+ T cells exhibit a Th1 phenotype and secrete IFN-γ and TNF-α [94]. These studies suggest that PCDs are mediated mainly by a CD8+ T-cell immune response toward an autoantigen recognized by onconeural Ab [64,81,95,96]. The cytotoxic lymphocytes appear to be aggressive toward cells presenting Cdr2 or Hu antigens and possibly induce cell death through one or more of the following mechanisms; 1) activation of TNF and FasL receptors, which triggers the apoptotic cell death cascade, and 2) excretion of granules that destroy cells through the actions of granzyme-B, perforin, and T-cell restricted intracellular antigen-1 (Figure 6) [81].
- Despite the above evidence suggesting the involvement of cell-mediated mechanisms in the development of PCDs, the transfer of lymphocytes from patients with anti-Yo Ab did not elicit any PC damage [97,98]. In addition, although all small cell lung carcinoma expresses the Hu antigen, and a clinically useful tumor immune response can be detected in up to 20% of the patients, the paraneoplastic neurologic syndrome is extremely rare, suggesting the robust tolerance to neural onconeural antigens in vivo [99]. Based on this notion, Blachère et al. [100] argued that CD8+ T cells that target intracellular antigens, in conjunction with CD4+ T cells, can induce tumor autoimmunity, but not neurologic autoimmunity. They proposed that 1) CD8+ T cells can only activate the peripheral immune response and 2) CD8+ T cells can break CNS tolerance to induce PCDs if they interact with both CD4+ T cells and autoantibodies [100]. This interaction between cell- and autoantibody-mediated mechanisms might provide the rationale for the design of new therapies that can target T and B cells using tacrolimus and rituximab, respectively [100].
- The following mechanisms can also underlie the break of CNS tolerance [101]: 1) structural modification of certain intracellular proteins, which stimulates the immune system, 2) death of cancer cells and release of intracellular antigens with subsequent exposure to the immune system, 3) persistence of the above autoimmune responses to neoplasm causes secretion of cytokines that elicit vascular hyperpermeability and infiltration of immune cells, and 4) simultaneous downregulation of regulatory T cells.
- Anti-GAD ataxia
- GAD is an enzyme that catalyzes the conversion of glutamate to GABA [102]. GAD has two isoforms: GAD65 and GAD67. The autoantibodies against the smaller isozyme are associated with CA [102]. Serum and CSF titers of anti-GAD65 Ab are higher in anti-GAD ataxia, at usually more than 10,000 U/mL (or 10 to 100-fold higher), compared to those of patients with type 1 diabetes mellitus (T1DM) [13,102]. The triggering factor of this autoimmunity is not clear at present. Anti-GAD ataxia can be associated with other types of IMCAs, such as PCD and GA [8]. While autoimmunity toward GAD65 affects the entire CNS, the cerebellum is one of the most vulnerable areas [102].
- The condition mostly affects women in their 60s, with subacute or chronic/insidious time course [8,102,103]. Some patients show evidence of other autoimmune conditions, such as T1DM, autoimmune thyroid diseases, and pernicious anemia [8,102,103]. Anti-GAD ataxia is sometimes associated with extracerebellar symptoms, including epilepsy, ophthalmoplegia and Stiff-Person syndrome [8,102,103]. The clinical course can be either subacute or chronic [103]. CSF studies sometimes show oligoclonal bands [8,102,103], whereas MRI shows normal or atrophic changes depending on the duration of illness [8,102,103]. The diagnosis is confirmed by the presence of high titers of intrathecally produced anti-GAD Ab [8,102,103].
- The serum level of anti-GAD antibodies in the context of the diagnosis of anti-GAD ataxia merits further consideration. There is currently no consensus on the threshold used for “low titer” and “high titer” levels of anti-GAD antibodies. In cases of anti-GAD Ab more than 2,000 U/mL (high titer), one can safely consider anti-GAD ataxia. However, titers in the range of 100–200 U/mL in an appropriate clinical/biological/neuroimaging context may be observed in immune ataxias. Some patients with CA and anti-GAD Ab less than 100 U/mL have been reported to show benefits to immunotherapies [102]. Anti-GAD Ab less than 100 U/mL may raise a suspicion of autoimmune diathesis and require therapy. It is also important to keep in mind that various laboratories use different techniques for the assays.
- The aim of any induction therapy should be to minimize progression of CA [9-11,102]. Thus, various immunotherapies, alone or in combination (more common), are used until remission [9-11,102]. Maintenance therapies are usually added to prevent any relapse [9-11,102]. Both the induction and maintenance therapies include corticosteroids, IVIg, immunosuppressants, plasmapheresis, and rituximab, either alone or in combination [9-11,102]. There appear to be no significant differences in the therapeutic benefits among the above immunotherapies [9-11,102]. The prognosis is better in the subacute type than in the chronic type of anti-GAD ataxia [103].
- The significance of anti-GAD65 has been a matter of debate [102,104]. Some researchers have argued that anti-GAD65 Ab have no pathogenic roles in the development of CAs based on the following reasons [105,106]: 1) anti-GAD65 Ab plays a pathogenic role in T1DM and various neurological conditions, such as Stiff-Person syndrome, 2) GAD65 is intracellularly located together with GABA transported with the cytosolic phase of GABA-containing vesicles, implying that autoantibodies do not have access to GAD65, and 3) lack of correlation between anti-GAD65 Ab titers and clinical symptoms [107]. However, recent physiological studies have provided substantial evidence for the pathogenic role of anti-GAD Ab [102,104]. It should be acknowledged that these studies did not rule out the secondary involvement of cell-mediated mechanisms.
- Studies using slice-tissue preparations showed that the addition of CSF IgGs from patients with anti-GAD ataxia to the medium resulted in presynaptic inhibition of GABAergic synapses between basket cells and PCs [108,109]. Furthermore, in in vivo preparations, application of CSF IgGs resulted in impairment of cerebellar-mediated modulations, as confirmed by various types of tasks [110-112]. Importantly, these pathogenic actions of IgGs from patients with anti-GAD ataxia were elicited by the binding of GA65 with anti-GAD65 Ab itself in an epitope-specific fashion: 1) these actions were abolished after the absorption of antiGAD65 Ab using recombinant GAD65 [113] and anti-GAD65 Ab elicited no actions in slices from GAD65 knockout mice where inhibitory transmissions were mediated compensatorily by GAD67 [112], 2) the b78 monoclonal Ab with epitope-specificity in CAs has pathogenic actions, whereas another monoclonal Ab has no such actions in T1DM [111,112], and 3) differences in neurological phenotype might also be related to epitope specificity [111,112]. Notably, low titer anti-GAD Ab has no pathogenic actions [102,104].
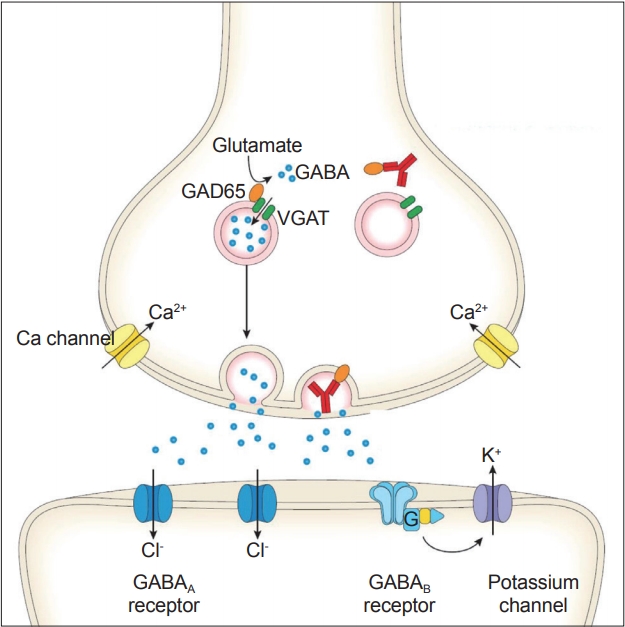
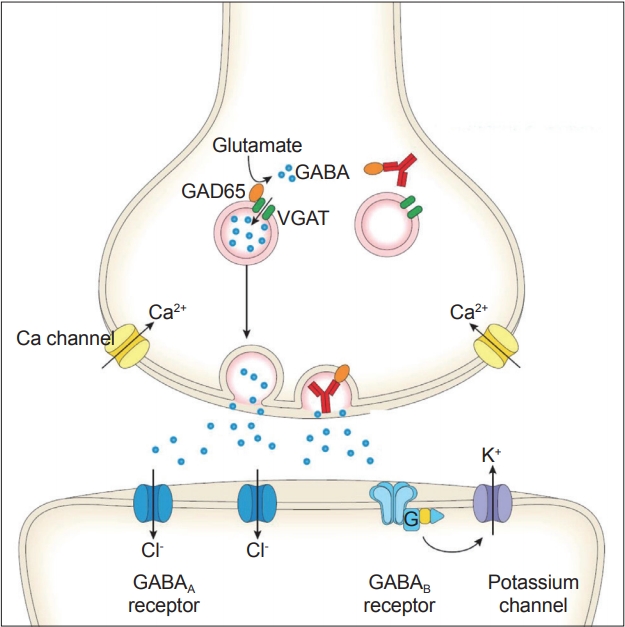
- Anti-GAD65 Ab is internalized, presumably during exocytosis or endocytosis [112,114]. Since exposure of GAD65 is assumed to be during exocytosis, anti-GAD65 could access GAD65 [102,104], although the exact mechanisms are still not clear. Anti-GAD65 Ab is reported to impair the association of GAD65 with vesicles, resulting in deficits in GABA packaging into vesicles and shuttling of vesicles to the release sites (Figure 7) [112].
- A decrease in GABA release attenuates the spill-over GABA-induced presynaptic inhibition on glutamate release from neighboring parallel fibers, resulting in a major imbalance between GABA and glutamate and excitotoxicity (Figure 8) [115]. Consistently, one autopsy study showed the complete loss of PCs [116].
- Previous studies showed that some patients respond well to immunotherapies and that the clinical improvement in CA correlates well with the fall in Ab titers [9-11], suggesting that Ab titers better reflect functional disorders rather than cell death. The peak CA might be influenced by secondary cell-mediated mechanisms.
- Taken together, the above experimental studies clearly show that anti-GAD65 Ab could serve as a trigger of CA (Figure 7). These studies also provide a generalized principle regarding antibody-induced neurological diseases; autoantibodies are polyclonal, and a cocktail of these polyclonal autoantibodies determines the clinical profile, depending on the proportions of pathogenic epitopes. In addition, pathomechanisms underlying the excitotoxicity could be generalized in other pathologies, such as ischemic diseases and degenerative CAs (Figure 8).
- Primary autoimmune cerebellar ataxia
- Although the clinical profile spectrum of PACA meets the features of IMCAs (e.g., subacute onset, autoimmune disease history, predominant gait ataxia, association of autoantibodies, and good benefits from immunotherapies), a subgroup of patients present with features that do not meet the criteria of the above common subtypes [8, 10,11,14,15,117,118]. Thus, the spectrum of PACA serves as an umbrella that covers heterogeneous etiologies [8,10,11,14,15,117,118]. Consistently, various types of less characterized autoantibodies are associated in some patients (Table 2), and immunohistochemical studies show a variety of staining patterns (60% of the patients) [14]. Despite the heterogeneity, patients with PACA sometimes show HLA type DQ2, which is observed in certain autoimmune diseases (CD, GA, T1DM, Stiff-Person syndrome, autoimmune thyroid disease and autoimmune polyendocrine syndromes), but not in healthy subjects [14,15].
- Many types of immunotherapies have been used in PACA. A small number of studies have reported the effectiveness of IVIg, prednisolone, plasmapheresis or rituximab in 9/19 patients at the early stage of PACA [119]. Progression to wheel-chair dependence was faster in patients with neuronal nuclear and/or cytoplasmic antibody than those positive for plasma membrane protein antibody [14], suggesting the need for the selection of therapeutic strategy based on the underlying autoimmune pathoetiology. In this regard, MR spectroscopy of the cerebellum could be an important tool in monitoring the response to immunotherapies [15,117,118].
PARTICULAR ISSUES IN COMMON ETIOLOGIES
Diagnosis and treatment
Autoimmune pathophysiological mechanisms
Myoclonic ataxia with refractory celiac disease
Diagnosis and treatment
Autoimmune pathophysiological mechanisms
Diagnosis and treatment
Autoimmune pathophysiological mechanisms
Diagnosis and treatment
Autoimmune pathophysiological mechanisms
Diagnosis and treatment
Autoimmune pathophysiological mechanisms
Epitope-specific actions
Internalization and dissociation of GAD65 with vesicles
From functional disorders to cell death
Clinical correlation
- This section focus on rare etiologies in IMCAs [117,118].
- Miller Fisher syndrome
- In 1956, Fisher120 proposed a new clinical entity of an acuteonset triad of ophthalmoplegia, ataxia and areflexia syndrome, which is currently termed Miller Fisher syndrome (MFS). MFS is assumed to be a representative disease of infection-triggered autoimmunity, similar to GBS [10]. A large-scale study involving 50 consecutive patients showed that viral infection (usually respiratory infection) or bacterial infection (usually Campylobacter jejuni) preceded the appearance of the triad manifestations, with a median interval of 8 days [121]. High serum titers of the specific marker anti-GQ1b Ab are found in up to 90% of patients with MFS, but not in those with other autoimmune neuropathies, such as GBS and chronic inflammatory demyelinating polyneuropathy. Anti-GQ1b Ab cross-reacts with epitopes present in the liposaccharide of MFS-associated Campylobacter jejuni strains, suggesting molecular mimicry [122].
- MFS shows self-limiting clinical course, thus requiring no immunotherapy [10]. One large-scale study showed full recovery within 6 months in all patients [121]. The speed of recovery and the final outcome were the same in patients who received corticosteroids, IVIg, or plasmapheresis and those who were treated conservatively [121]. The good prognosis suggests that any autoimmune-mediated impairment in MFS is reversible. It remains to be determined whether the autoimmune response functionally attacks peripheral or central structures. Based on the findings of GQ1b expression in large-diameter dorsal ganglion neurons [8] and the presence of enhanced lesions in the spinocerebellar tracts at the lower medulla on MRI [123], several researchers have argued for the presence of peripheral lesions in MFS. Moreover, cerebellar involvement has been confirmed more directly in a number of recent studies. One study using FDG-PET showed hypermetabolism in the cerebellum and brainstem [124], and another study using MR spectroscopy showed abnormal signals in the cerebellum during the course of the illness [125]. Furthermore, immunocytochemical studies using sera of MFS patients confirmed the presence of autoantibodies to antigens in the molecular layer [126,127].
- Recently established clinical entities
- Dipeptidyl-peptidase-like protein-6 (DPPX), an auxiliary subunit of the Kv4.2 potassium channel [128-130], and contactin-associated protein-like 2 (Caspr2), a protein associated with the Kv1 potassium channel [131,132], can be targets of autoimmunity. Since potassium channels are distributed throughout the CNS, CAs are one of the diverse features of these etiologies. Patients with anti-DPPX Ab show multifocal neurological symptoms such as agitation, mild confusion, hyperekplexia, myoclonus, trunk stiffness and CAs, which are attributed to the hyperexcitability induced by potassium channel deficits [128-130]. Recently, CAs with myoclonus were also reported as the sole manifestation of this entity [10]. Additionally, encephalitis associated with anti-Caspr2 Ab is defined as ≥ 3 core manifestations, including encephalic signs, CAs, peripheral nerve hyperexcitability, dysautonomia, neuropathic pain, insomnia, and weight loss [131]. Notably, stereotypical episodes of paroxysmal CAs (mimicking episodic ataxias) are found in some patients with anti-Caspr2 Ab [132]. The gait imbalance, limb ataxia, and slurred speech usually last a few minutes to a few days and usually improve following immunotherapy. These two novel entities suggest that the cerebellum is one of the vulnerable areas in patients with potassium channel dysfunction.
- Myelin-associated glycoprotein (MAG) plays an important role in the maintenance of the myelin sheath, not only in Schwann cells but also in the CNS [133]. The association of anti-MAG Ab with distal demyelinating neuropathy is well established in patients with monoclonal gammopathy of unknown significance [134]. Recently, CA associated with anti-MAG Ab was described in five patients [134]. All patients had IgM gammopathy, and four of the five showed clinically evident neuropathy. MR spectroscopy showed cerebellar involvements, which diminished after improvement of CAs with rituximab [134].
- In contrast to these autoantibody-related etiologies, chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids (CLIPPERS) is characterized by marked perivascular T cell inflammation [135,136]. The lymphocytes, mainly CD4-dominant lymphocytes, infiltrate the perivascular area and more diffusely in the white matter of the pons and adjacent rhombencephalic structures, such as the cerebellar peduncles, cerebellum, medulla, and the midbrain [135,136]. The perivascular infiltration appears as characteristic gadolinium enhancement on MRI, with multiple “punctate” and/or “curvilinear” gadolinium enhancing lesions, resulting in “peppering” of the pons with or without peripontine lesions [135,136]. The conditions affect mainly men in their 50s [135,136]. The patients show subacute onset of the combination of brainstem symptoms and CAs, which include pancerebellar ataxias, dysarthria, dysphagia, dysgeusia, oculomotor abnormalities, altered facial sensation, facial nerve palsy, vertigo, pyramidal signs and sensory disorders [135,136]. CLIPPERS is characterized by responsiveness to corticosteroids [135,136]. The initial treatment is intravenous methyl prednisolone followed by maintenance immunotherapy using the combination of oral prednisolone and corticosteroid-sparing immunosuppressants [135,136]. Since the clinical course seems to be relapsing–remitting in nature, long-term maintenance therapy is required [136].
OTHER RARE ETIOLOGIES IN IMCAs
- IMCAs include diverse etiologies, suggesting that the cerebellum can be the target of many types of autoimmune responses with different pathophysiological mechanisms. The identification of new clinical entities (anti-DPPX ataxia, anti-Caspr2 ataxia, anti-MAG ataxia, and CLIPPERS) suggests that further novel entities are likely to be discovered in the near future. IMCAs of certain etiologies respond well to immunotherapy at least in the early stage, while the cerebellar reserve is preserved. Thus, clinicians should identify therapeutic opportunities early [17]. Conversely, a subgroup of etiologies shows poor prognosis, thus requiring a future breakthrough in therapeutic strategies [137]. Further advances in our understanding of these complex immune mechanisms are important in dictating future immunotherapies.
CONCLUSION
-
Conflicts of Interest
The authors have no financial conflicts of interest.
-
Author Contributions
Conceptualization: Hiroshi Mitoma. Data curation: Hiroshi Mitoma. Formal analysis: Hiroshi Mitoma. Investigation: Hiroshi Mitoma. Project administration: Hiroshi Mitoma, Mario Manto. Resources: Marios Hadjivassiliou. Supervision: Mario Manto, Marios Hadjivassiliou. Validation: all authors. Writing—original draft: Hiroshi Mitoma. Writing—review&editing: Mario Manto, Marios Hadjivassiliou.
Notes
- None
Acknowledgments


| Autoimmunity that mainly targets the cerebellum* or its related structures | |
| Cerebellar autoimmunity triggered by another disease or condition | |
| Gluten ataxia | (gluten sensitivity) |
| Postinfectious cerebellitis | (infection) |
| Miller Fisher syndrome | (infection) |
| Opsoclonus myoclonus syndrome† | (infection, neoplasm) |
| Paraneoplastic cerebellar degenerations | (neoplasm) |
| Cerebellar autoimmunity not triggered by another disease or condition | |
| Anti-GAD ataxia‡ | |
| Primary autoimmune cerebellar ataxia | |
| Others | |
* when cerebellar ataxias are the sole or main symptoms, the cerebellum is presumed to be the main target of autoimmunity. The term IMCAs is reserved for mainly pure or primarily cerebellar ataxia, thus excluding more widespread autoimmune damage,
† opsoclonus myoclonus syndrome also includes idiopathic type,
‡ anti-GAD ataxia occurs in other autoimmune backgrounds, such as neoplasm.
IMCAs: immune-mediated cerebellar ataxias. GAD: glutamic acid decarboxylase. Adapted from Mitoma et al. Cerebellum 2016;15:213-232, under the terms of the Creative Commons CC BY license. [8]
Unknown conditions might be PACA. IMCAs: immune-mediated cerebellar ataxias, PCDs: paraneoplastic cerebellar degenerations, CA: cerebellar ataxia, PIC: postinfectious cerebellitis, OMS: opsoclonus myoclonus syndrome, PACA: primary autoimmune cerebellar ataxia, TG: transglutaminase, VGCC: voltage gated calcium channel, DPPX: dipeptidyl-peptidase-like protein 6, LGI1: leucine-rich glioma-inactivated 1, CASPR2: contactin-associated protein-like 2, mGluR1: metabotropic glutamate receptor, GAD65: glutamic acid decarboxylase 65, MAG: myelin-associated glycoprotein, ZIC4: zinc finger protein of the cerebellum 4, PCA2: purkinje cell antibody 2, GluRδ2: glutamate receptor delta2, PKCγ: protein kinase C gamma, Ca/ARHGAP26: Ca/Rho GTPase-activating protein 26, SOX1: sex determining region Y-related high-mobility group box 1, CARP VIII: carbonic anhydrase-related protein VIII, Sj/ITPR-1: Sj/inositol 1,4,5-trisphosphate receptor 1, Nb/AP3B2: Nb/adaptor complex 3 B2. Adapted from Mitoma et al. MedLink Neurology 2019 Aug 23 (www.medlink.com), with permission of MedLink. [11]
EBV: Epstein-Barr virus, HLA: human leukocyte antigen, WBC: white blood cell, TG: transglutaminase, VGCC: voltage gated calcium channel, CASPR2: contactin-associated protein-like 2, mGluR: metabotropic glutamate receptor, GAD65: glutamic acid decarboxylase 65, ZIC4: zinc finger protein of the cerebellum 4, PCA2: purkinje cell antibody 2, GluRδ2: glutamate receptor delta2, PKCγ: protein kinase C gamma, Ca/ARHGAP26: Ca/Rho GTPase-activating protein 26, SOX1: sex determining region Y-related high-mobility group box 1, CARP VIII: carbonic anhydrase-related protein VIII. Adapted from Mitoma et al. MedLink Neurology 2019 Aug 23 (www.medlink.com), with permission of MedLink. [11]
- 1. Manto M, Bower JM, Conforto AB, Delgado-García JM, da Guarda SN, Gerwig M, et al. Consensus paper: roles of the cerebellum in motor control—The diversity of ideas on cerebellar involvement in movement. Cerebellum 2012;11:457–487.ArticlePubMedPMC
- 2. Schmahmann JD, Caplan D. Cognition, emotion and the cerebellum. Brain 2006;129:290–292.ArticlePubMed
- 3. Charcot JM. Séance du 14mars. CR Soc Biol (Paris) 1868;20:13.
- 4. Brouwer B. Beitrag zur kenntnis der chronischen diffusen kleinhirnerkranghkungen. Neurol Centralbl 1919;38:674–682.
- 5. Greenlee JE, Brashear HR. Antibodies to cerebellar Purkinje cells in patients with paraneoplastic cerebellar degeneration and ovarian carcinoma. Ann Neurol 1983;14:609–613.ArticlePubMed
- 6. Ducray F, Demarquay G, Graus F, Decullier E, Antoine JC, Giometto B, et al. Seronegative paraneoplastic cerebellar degeneration: the PNS Euronetwork experience. Eur J Neurol 2014;21:731–735.ArticlePubMed
- 7. Hadjivassiliou M. Immune-mediated acquired ataxias. Handb Clin Neurol 2012;103:189–199.ArticlePubMed
- 8. Mitoma H, Adhikari K, Aeschlimann D, Chattopadhyay P, Hadjivassiliou M, Hampe CS, et al. Consensus paper: neuroimmune mechanisms of cerebellar ataxias. Cerebellum 2016;15:213–232.ArticlePubMed
- 9. Mitoma H, Hadjivassiliou M, Honnorat J. Guidelines for treatment of immune-mediated cerebellar ataxias. Cerebellum Ataxias 2015;2:14.ArticlePubMedPMC
- 10. Hadjivassiliou M, Mitoma H, Manto M. Autoimmune ataxia. In: Mitoma H, Manto M, editors. Neuroimmune Diseases: From Cells to the Living Brain. Cham, Switzerland: Springer Nature; 2019:599–620.
- 11. Mitoma H, Manto M, Hadjivassiliou M. Nonparaneoplastic autoimmune cerebellar ataxias. MedLink Neurology; 2019 Aug. 23. [upadated 2020 Aug 28]. San Diego: MedLink Corporation. Available from: https://www.medlink.com/article/nonparaneoplastic_autoimmune_cerebellar_ataxias.
- 12. Hadjivassiliou M, Grünewald RA, Chattopadhyay AK, Davies-Jones GA, Gibson A, Jarratt JA, et al. Clinical, radiological, neurophysiological, and neuropathological characteristics of gluten ataxia. Lancet 1998;352:1582–1585.ArticlePubMed
- 13. Honnorat J, Saiz A, Giometto B, Vincent A, Brieva L, de Andres C, et al. Cerebellar ataxia with anti-glutamic acid decarboxylase antibodies: study of 14 patients. Arch Neurol 2001;58:225–230.ArticlePubMed
- 14. Hadjivassiliou M, Boscolo S, Tongiorgi E, Grünewald RA, Sharrack B, Sanders DS, et al. Cerebellar ataxia as a possible organ-specific autoimmune disease. Mov Disord 2008;23:1370–1377.ArticlePubMed
- 15. Hadjivassiliou M, Graus F, Honnorat J, Jarius S, Titulaer M, Manto M, et al. Diagnostic criteria for primary autoimmune cerebellar ataxia–guidelines from an International Task Force on Immune-Mediated Cerebellar Ataxias. Cerebellum 2020;19:605–610.ArticlePubMedPMC
- 16. Mitoma H, Manto M. The physiological basis of therapies for cerebellar ataxias. Ther Adv Neurol Disord 2016;9:396–413.ArticlePubMedPMC
- 17. Mitoma H, Manto M, Hampe CS. Time is cerebellum. Cerebellum 2018;17:387–391.ArticlePubMedPMC
- 18. In: Mitoma M, Manto M, editors. Neuroimmune Diseases: From Cells to Living Brains. Cham, Switzerland: Springer Nature; 2019.
- 19. Hadjivassiliou M, Martindale J, Shanmugarajah P, Grünewald RA, Sarrigiannis PG, Beauchamp N, et al. Causes of progressive cerebellar ataxia: prospective evaluation of 1500 patients. J Neurol Neurosurg Psychiatry 2017;88:301–309.ArticlePubMed
- 20. Gebus O, Montaut S, Monga B, Wirth T, Cheraud C, Alves Do Rego C, et al. Deciphering the causes of sporadic late-onset cerebellar ataxias: a prospective study with implications for diagnostic work. J Neurol 2017;264:1118–1126.ArticlePubMed
- 21. Kim JS, Kwon S, Ki CS, Youn J, Cho JW. The etiologies of chronic progressive cerebellar ataxia in a Korean population. J Clin Neurol 2018;14:374–380.ArticlePubMedPMC
- 22. Nanri K, Okuma M, Sato S, Yoneda M, Taguchi T, Mitoma H, et al. Prevalence of autoantibodies and the efficacy of immunotherapy for autoimmune cerebellar ataxia. Intern Med 2016;55:449–454.ArticlePubMed
- 23. Guan WJ, Liu XJ, Tang BS, Liu YT, Zhou Y, Jiang H, et al. Gluten ataxia of sporadic and hereditary cerebellar ataxia in patients from mainland China. Neurol India 2013;61:226–230.ArticlePubMed
- 24. Muñiz-Castrillo S, Honnorat J. Paraneoplastic neurological syndromes. Mitoma H, Manto M. Neuroimmune Diseases: From Cells to the Living Brain. Cham, Switzerland: Springer Nature; 2019:439–485.
- 25. Jarius S, Wildemann B. ‘Medusa-head ataxia’: the expanding spectrum of Purkinje cell antibodies in autoimmune cerebellar ataxia. Part 1: antimGluR1, anti-Homer-3, anti-Sj/ITPR1 and anti-CARP VIII. J Neuroinflammation 2015;12:166.ArticlePubMedPMC
- 26. Jarius S, Wildemann B. ‘Medusa head ataxia’: the expanding spectrum of Purkinje cell antibodies in autoimmune cerebellar ataxia. Part 2: anti-PKC-gamma, anti-GluR-delta2, anti-Ca/ARHGAP26 and anti-VGCC. J Neuroinflammation 2015;12:167.ArticlePubMedPMC
- 27. Jarius S, Wildemann B. ‘Medusa head ataxia’: the expanding spectrum of Purkinje cell antibodies in autoimmune cerebellar ataxia. Part 3: antiYo/CDR2, anti-Nb/AP3B2, PCA-2, anti-Tr/DNER, other antibodies, diagnostic pitfalls, summary and outlook. J Neuroinflammation 2015;12:168.ArticlePubMedPMC
- 28. Fukuda T, Motomura M, Nakao Y, Shiraishi H, Yoshimura T, Iwanaga K, et al. Reduction of P/Q-type calcium channels in the postmortem cerebellum of paraneoplastic cerebellar degeneration with Lambert-Eaton myasthenic syndrome. Ann Neurol 2003;53:21–28.ArticlePubMed
- 29. Liao YJ, Safa P, Chen YR, Sobel RA, Boyden ES, Tsien RW. Anti-Ca2+ channel antibody attenuates Ca2+ currents and mimics cerebellar ataxia in vivo. Proc Natl Acad Sci U S A 2008;105:2705–2710.ArticlePubMedPMC
- 30. Piepgras J, Höltje M, Michel K, Li Q, Otto C, Drenckhahn C, et al. AntiDPPX encephalitis: pathogenic effects of antibodies on gut and brain neurons. Neurology 2015;85:890–897.ArticlePubMedPMC
- 31. Petit-Pedrol M, Sell J, Planagumà J, Mannara F, Radosevic M, Haselmann H, et al. LGI1 antibodies alter Kv1.1 and AMPA receptors changing synaptic excitability, plasticity and memory. Brain 2018;141:3144–3159.ArticlePubMedPMC
- 32. Saint-Martin M, Pieters A, Déchelotte B, Malleval C, Pinatel D, Pascual O, et al. Impact of anti-CASPR2 autoantibodies from patients with autoimmune encephalitis on CASPR2/TAG-1 interaction and Kv1 expression. J Autoimmun 2019;103:102284.ArticlePubMed
- 33. Coesmans M, Smitt PA, Linden DJ, Shigemoto R, Hirano T, Yamakawa Y, et al. Mechanisms underlying cerebellar motor deficits due to mGluR1- autoantibodies. Ann Neurol 2003;53:325–336.ArticlePubMed
- 34. Hampe CS. Significance of autoantibodies. In: Mitoma H, Manto M, editors. Neuroimmune Diseases: From Cells to the Living Brain. Cham, Switzerland: Springer Nature; 2019:109–142.
- 35. Baldarçara L, Currie S, Hadjivassiliou M, Hoggard N, Jack A, Jackowski AP, et al. Consensus paper: radiological biomarkers of cerebellar diseases. Cerebellum 2015;14:175–196.ArticlePubMedPMC
- 36. Hadjivassiliou M, Sanders DS, Grünewald RA, Woodroofe N, Boscolo S, Aeschlimann D. Gluten sensitivity: from gut to brain. Lancet Neurol 2010;9:318–330.ArticlePubMed
- 37. Hadjivassiliou M, Aeschlimann P, Sanders DS, Mäki M, Kaukinen K, Grünewald RA, et al. Transglutaminase 6 antibodies in the diagnosis of gluten ataxia. Neurology 2013;80:1740–1745.ArticlePubMed
- 38. Hadjivassiliou M, Davies-Jones GAB, Sanders DS, Grünewald RA. Dietary treatment of gluten ataxia. J Neurol Neurosurg Psychiatry 2003;74:1221–1224.ArticlePubMedPMC
- 39. Souayah N, Chin RL, Brannagan TH, Latov N, Green PH, Kokoszka A, et al. Effect of intravenous immunoglobulin on cerebellar ataxia and neuropathic pain associated with celiac disease. Eur J Neurol 2008;15:1300–1303.ArticlePubMed
- 40. Hadjivassiliou M, Grünewald RA, Davies-Jones GA. Gluten sensitivity as a neurological illness. J Neurol Neurosurg Psychiatry 2002;72:560–563.ArticlePubMedPMC
- 41. Hadjivassiliou M, Sanders DS, Woodroofe N, Williamson C, Grünewald RA. Gluten ataxia. Cerebellum 2008;7:494–498.ArticlePubMed
- 42. Hadjivassiliou M, Grünewald RA, Sanders DS, Shanmugarajah P, Hoggard N. Effect of gluten-free diet on cerebellar MR spectroscopy in gluten ataxia. Neurology 2017;89:705–709.ArticlePubMed
- 43. Schuppan D, Junker Y, Barisani D. Celiac disease: from pathogenesis to novel therapies. Gastroenterology 2009;137:1912–1933.ArticlePubMed
- 44. Boscolo S, Lorenzon A, Sblattero D, Florian F, Stebel M, Marzari R, et al. Anti transglutaminase antibodies cause ataxia in mice. PLoS One 2010;5:e9698.ArticlePubMedPMC
- 45. Cervio E, Volta U, Verri M, Boschi F, Pastoris O, Granito A, et al. Sera of patients with celiac disease and neurologic disorders evoke a mitochondrial-dependent apoptosis in vitro. Gastroenterology 2007;133:195–206.ArticlePubMed
- 46. Iversen R, Di Niro R, Stamnaes J, Lundin KE, Wilson PC, Sollid LM. Transglutaminase 2-specific autoantibodies in celiac disease target clustered, N-terminal epitopes not displayed on the surface of cells. J Immunol 2013;190:5981–5991.ArticlePubMedPMC
- 47. Wang JL, Yang X, Xia K, Hu ZM, Weng L, Jin X, et al. TGM6 identified as a novel causative gene of spinocerebellar ataxias using exome sequencing. Brain 2010;133:3510–3518.ArticlePubMed
- 48. Sarrigiannis PG, Hoggard N, Aeschlimann D, Sanders DS, Grünewald RA, Unwin ZC, et al. Myoclonus ataxia and refractory coeliac disease. Cerebellum Ataxias 2014;1:11.ArticlePubMedPMC
- 49. Shibasaki H, Hallett M. Electrophysiological studies of myoclonus. Muscle Nerve 2005;31:157–174.ArticlePubMed
- 50. Blumkin L, Pranzatelli MR. Acquired ataxias, infectious and para-infectious. Handb Clin Neurol 2012;103:137–146.ArticlePubMed
- 51. Sawaishi Y, Takada G. Acute cerebellitis. Cerebellum 2002;1:223–228.ArticlePubMed
- 52. Sivaswamy L. Approach to acute ataxia in childhood: diagnosis and evaluation. Pediatr Ann 2014;43:153–159.ArticlePubMed
- 53. Connolly AM, Dodson WE, Prensky AL, Rust RS. Course and outcome of acute cerebellar ataxia. Ann Neurol 1994;35:673–679.ArticlePubMed
- 54. Hirai H, Launey T, Mikawa S, Torashima T, Yanagihara D, Kasaura T, et al. New role of delta2-glutamate receptors in AMPA receptor trafficking and cerebellar function. Nat Neurosci 2003;6:869–876.ArticlePubMed
- 55. Armangué T, Sabater L, Torres-Vega E, Martínez-Hernández E, Ariño H, Petit-Pedrol M, et al. Clinical and immunological features of opsoclonus-myoclonus syndrome in the era of neuronal cell surface antibodies. JAMA Neurol 2016;73:417–424.ArticlePubMedPMC
- 56. Bataller L, Graus F, Saiz A, Vilchez JJ; Spanish Opsoclonus-Myoclonus Study Group. Clinical outcome in adult onset idiopathic or paraneoplastic opsoclonus-myoclonus. Brain 2001;124:437–443.ArticlePubMed
- 57. Klaas JP, Ahlskog JE, Pittock SJ, Matsumoto JY, Aksamit AJ, Bartleson JD, et al. Adult-onset opsoclonus-myoclonus syndrome. Arch Neurol 2012;69:1598–1607.ArticlePubMed
- 58. Pranzatelli MR, Travelstead AL, Tate ED, Allison TJ, Moticka EJ, Franz DN, et al. B- and T-cell markers in opsoclonus-myoclonus syndrome: immunophenotyping of CSF lymphocytes. Neurology 2004;62:1526–1532.ArticlePubMed
- 59. Pranzatelli MR, Tate ED, Swan JA, Travelstead AL, Colliver JA, Verhulst SJ, et al. B cell depletion therapy for new-onset opsoclonus-myoclonus. Mov Disord 2010;25:238–242.ArticlePubMed
- 60. Young CA, MacKenzie JM, Chadwick DW, Williams IR. Opsoclonusmyoclonus syndrome: an autopsy study of three cases. Eur J Med 1993;2:239–241.PubMed
- 61. Mitoma H, Orimo S, Sodeyama N, Tamaki M. Paraneoplastic opsoclonus-myoclonus syndrome and neurofibrosarcoma. Eur Neurol 1996;36:322–324.
- 62. Wong AM, Musallam S, Tomlinson RD, Shannon P, Sharpe JA. Opsoclonus in three dimensions: oculographic, neuropathologic and modelling correlates. J Neurol Sci 2001;189:71–81.ArticlePubMed
- 63. Chekroud AM, Anand G, Yong J, Pike M, Bridge H. Altered functional brain connectivity in children and young people with opsoclonus-myoclonus syndrome. Dev Med Child Neurol 2017;59:98–104.ArticlePubMed
- 64. Dalmau J, Rosenfeld MR. Paraneoplastic syndromes of the CNS. Lancet Neurol 2008;7:327–340.ArticlePubMedPMC
- 65. Graus F, Delattre JY, Antoine JC, Dalmau J, Giometto B, Grisold W, et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry 2004;75:1135–1140.ArticlePubMedPMC
- 66. Höftberger R, Rosenfeld MR, Dalmau J. Update on neurological paraneoplastic syndromes. Curr Opin Oncol 2015;27:489–495.ArticlePubMedPMC
- 67. Demarquay G, Honnorat J. Clinical presentation of immune-mediated cerebellar ataxia. Rev Neurol (Paris) 2011;167:408–417.ArticlePubMed
- 68. Voltz R. Paraneoplastic neurological syndromes: an update on diagnosis, pathogenesis, and therapy. Lancet Neurol 2002;1:294–305.ArticlePubMed
- 69. Shams’ili S, Grefkens J, de Leeuw B, van den Bent M, Hooijkaas H, van der Holt B, et al. Paraneoplastic cerebellar degeneration associated with antineuronal antibodies: analysis of 50 patients. Brain 2003;126:1409–1418.ArticlePubMed
- 70. Peterson K, Rosenblum MK, Kotanides H, Posner JB. Paraneoplastic cerebellar degeneration. I. A clinical analysis of 55 anti-Yo antibody-positive patients. Neurology 1992;42:1931–1937.ArticlePubMed
- 71. Rojas I, Graus F, Keime-Guibert F, Reñé R, Delattre JY, Ramón JM, et al. Long-term clinical outcome of paraneoplastic cerebellar degeneration and anti-Yo antibodies. Neurology 2000;55:713–715.ArticlePubMed
- 72. Candler PM, Hart PE, Barnett M, Weil R, Rees JH. A follow up study of patients with paraneoplastic neurological disease in the United Kingdom. J Neurol Neurosurg Psychiatry 2004;75:1411–1415.ArticlePubMedPMC
- 73. Keime-Guibert F, Graus F, Fleury A, René R, Honnorat J, Broet P, et al. Treatment of paraneoplastic neurological syndromes with antineuronal antibodies (Anti-Hu, anti-Yo) with a combination of immunoglobulins, cyclophosphamide, and methylprednisolone. J Neurol Neurosurg Psychiatry 2000;68:479–482.ArticlePubMedPMC
- 74. Verschuuren J, Chuang L, Rosenblum MK, Lieberman F, Pryor A, Posner JB, et al. Inflammatory infiltrates and complete absence of Purkinje cells in anti-Yo-associated paraneoplastic cerebellar degeneration. Acta Neuropathol 1996;91:519–525.ArticlePubMed
- 75. Giometto B, Marchiori GC, Nicolao P, Scaravilli T, Lion A, Bardin PG, et al. Sub-acute cerebellar degeneration with anti-Yo autoantibodies: immunohistochemical analysis of the immune reaction in the central nervous system. Neuropathol Appl Neurobiol 1997;23:468–474.ArticlePubMed
- 76. Storstein A, Krossnes BK, Vedeler CA. Morphological and immunohistochemical characterization of paraneoplastic cerebellar degeneration associated with Yo antibodies. Acta Neurol Scand 2009;120:64–67.ArticlePubMed
- 77. Aboul-Enein F, Höftberger R, Buxhofer-Ausch V, Drlicek M, Lassmann H, Budka H, et al. Neocortical neurones may be targeted by immune attack in anti-Yo paraneoplastic syndrome. Neuropathol Appl Neurobiol 2008;34:248–252.ArticlePubMed
- 78. Aye MM, Kasai T, Tashiro Y, Xing HQ, Shirahama H, Mitsuda M, et al. CD8 positive T-cell infiltration in the dentate nucleus of paraneoplastic cerebellar degeneration. J Neuroimmunol 2009;208:136–140.ArticlePubMed
- 79. Brain WR, Daniel PM, Greenfield JG. Subacute cortical cerebellar degeneration and its relation to carcinoma. J Neurol Neurosurg Psychiatry 1951;14:59–75.ArticlePubMedPMC
- 80. Brock S, Ellison D, Frankel J, Davis C, Illidge T. Anti-Yo antibody-positive cerebellar degeneration associated with endometrial carcinoma: case report and review of the literature. Clin Oncol (R Coll Radiol) 2001;13:476–479.ArticlePubMed
- 81. Zaborowski MP, Michalak S. Cell-mediated immune responses in paraneoplastic neurological syndromes. Clin Dev Immunol 2013;2013:630602.ArticlePubMedPMC
- 82. Okano HJ, Park WY, Corradi JP, Darnell RB. The cytoplasmic Purkinje onconeural antigen cdr2 down-regulates c-Myc function: implications for neuronal and tumor cell survival. Genes Dev 1999;13:2087–2097.ArticlePubMedPMC
- 83. Sakai K, Kitagawa Y, Saiki S, Saiki M, Hirose G. Effect of a paraneoplastic cerebellar degeneration-associated neural protein on B-myb promoter activity. Neurobiol Dis 2004;15:529–533.ArticlePubMed
- 84. Tanaka K, Ding X, Tanaka M. Effects of antineuronal antibodies from patients with paraneoplastic neurological syndrome on primary-cultured neurons. J Neurol Sci 2004;217:25–30.ArticlePubMed
- 85. Lancaster E, Dalmau J. Neuronal autoantigens--pathogenesis, associated disorders and antibody testing. Nat Rev Neurol 2012;8:380–390.ArticlePubMedPMC
- 86. Tanaka K, Tanaka M, Igarashi S, Onodera O, Miyatake T, Tsuji S. Trial to establish an animal model of paraneoplastic cerebellar degeneration with anti-Yo antibody. 2. Passive transfer of murine mononuclear cells activated with recombinant Yo protein to paraneoplastic cerebellar degeneration lymphocytes in severe combined immunodeficiency mice. Clin Neurol Neurosurg 1995;97:101–105.ArticlePubMed
- 87. Sillevis Smitt PA, Manley GT, Posner JB. Immunization with the paraneoplastic encephalomyelitis antigen HuD does not cause neurologic disease in mice. Neurology 1995;45:1873–1878.ArticlePubMed
- 88. Carpentier AF, Rosenfeld MR, Delattre JY, Whalen RG, Posner JB, Dalmau J. DNA vaccination with HuD inhibits growth of a neuroblastoma in mice. Clin Cancer Res 1998;4:2819–2824.PubMed
- 89. Benyahia B, Liblau R, Merle-Béral H, Tourani JM, Dalmau J, Delattre JY. Cell-mediated autoimmunity in paraneoplastic neurological syndromes with anti-Hu antibodies. Ann Neurol 1999;45:162–167.ArticlePubMed
- 90. Albert ML, Austin LM, Darnell RB. Detection and treatment of activated T cells in the cerebrospinal fluid of patients with paraneoplastic cerebellar degeneration. Ann Neurol 2000;47:9–17.ArticlePubMed
- 91. Rousseau A, Benyahia B, Dalmau J, Connan F, Guillet JG, Delattre JY, et al. T cell response to Hu-D peptides in patients with anti-Hu syndrome. J Neurooncol 2005;71:231–236.ArticlePubMedPMC
- 92. Plonquet A, Gherardi RK, Créange A, Antoine JC, Benyahia B, Grisold W, et al. Oligoclonal T-cells in blood and target tissues of patients with anti-Hu syndrome. J Neuroimmunol 2002;122:100–105.ArticlePubMed
- 93. Wanschitz J, Hainfellner JA, Kristoferitsch W, Drlicek M, Budka H. Ganglionitis in paraneoplastic subacute sensory neuronopathy: a morphologic study. Neurology 1997;49:1156–1159.ArticlePubMed
- 94. Gebauer C, Pignolet B, Yshii L, Mauré E, Bauer J, Liblau R. CD4+ and CD8+ T cells are both needed to induce paraneoplastic neurological disease in a mouse model. Oncoimmunology 2016;6:e1260212.ArticlePubMedPMC
- 95. Roberts WK, Deluca IJ, Thomas A, Fak J, Williams T, Buckley N, et al. Patients with lung cancer and paraneoplastic Hu syndrome harbor HuDspecific type 2 CD8+ T cells. J Clin Invest 2009;119:2042–2051.ArticlePubMedPMC
- 96. Albert ML, Darnell JC, Bender A, Francisco LM, Bhardwaj N, Darnell RB. Tumor-specific killer cells in paraneoplastic cerebellar degeneration. Nat Med 1998;4:1321–1324.ArticlePubMed
- 97. Venkatraman A, Opal P. Paraneoplastic cerebellar degeneration with anti-Yo antibodies - a review. Ann Clin Transl Neurol 2016;3:655–663.ArticlePubMedPMC
- 98. Sakai K, Mitchell DJ, Tsukamoto T, Steinman L. Isolation of a complementary DNA clone encoding an autoantigen recognized by an antineuronal cell antibody from a patient with paraneoplastic cerebellar degeneration. Ann Neurol 1990;28:692–698.ArticlePubMed
- 99. DeLuca I, Blachère NE, Santomasso B, Darnell RB. Tolerance to the neuron-specific paraneoplastic HuD antigen. PLoS One 2009;4:e5739.ArticlePubMedPMC
- 100. Blachère NE, Orange DE, Santomasso BD, Doerner J, Foo PK, Herre M, et al. T cells targeting a neuronal paraneoplastic antigen mediate tumor rejection and trigger CNS autoimmunity with humoral activation. Eur J Immunol 2014;44:3240–3251.ArticlePubMedPMC
- 101. Zaenker P, Gray ES, Ziman MR. Autoantibody production in cancer—the humoral immune response toward autologous antigens in cancer patients. Autoimmun Rev 2016;15:477–483.ArticlePubMed
- 102. Mitoma H, Manto M, Hampe CS. Pathogenic roles of glutamic acid decarboxylase 65 autoantibodies in cerebellar ataxias. J Immunol Res 2017;2017:2913297.ArticlePubMedPMC
- 103. Ariño H, Gresa-Arribas N, Blanco Y, Martínez-Hernández E, Sabater L, Petit-Pedrol M, et al. Cerebellar ataxia and glutamic acid decarboxylase antibodies: immunologic profile and long-term effect of immunotherapy. JAMA Neurol 2014;71:1009–1016.ArticlePubMedPMC
- 104. Manto M, Mitoma H, Hampe CS. Anti-GAD antibodies and the cerebellum: where do we stand? Cerebellum 2019;18:153–156.ArticlePubMed
- 105. Gresa-Arribas N, Ariño H, Martínez-Hernández E, Petit-Pedrol M, Sabater L, Saiz A, et al. Antibodies to inhibitory synaptic proteins in neurological syndromes associated with glutamic acid decarboxylase autoimmunity. PLoS One 2015;10:e0121364.ArticlePubMedPMC
- 106. Balint B, Bhatia KP. Stiff person syndrome and other immune-mediated movement disorders - new insights. Curr Opin Neurol 2016;29:496–506.ArticlePubMed
- 107. Rakocevic G, Raju R, Dalakas MC. Anti-glutamic acid decarboxylase antibodies in the serum and cerebrospinal fluid of patients with stiffperson syndrome: correlation with clinical severity. Arch Neurol 2004;61:902–904.ArticlePubMed
- 108. Ishida K, Mitoma H, Song SY, Uchihara T, Inaba A, Eguchi S, et al. Selective suppression of cerebellar GABAergic transmission by an autoantibody to glutamic acid decarboxylase. Ann Neurol 1999;46:263–267.ArticlePubMed
- 109. Mitoma H, Song SY, Ishida K, Yamakuni T, Kobayashi T, Mizusawa H. Presynaptic impairment of cerebellar inhibitory synapses by an autoantibody to glutamate decarboxylase. J Neurol Sci 2000;175:40–44.ArticlePubMed
- 110. Manto MU, Laute MA, Aguera M, Rogemond V, Pandolfo M, Honnorat J. Effects of anti-glutamic acid decarboxylase antibodies associated with neurological diseases. Ann Neurol 2007;61:544–551.ArticlePubMed
- 111. Manto MU, Hampe CS, Rogemond V, Honnorat J. Respective implications of glutamate decarboxylase antibodies in stiff person syndrome and cerebellar ataxia. Orphanet J Rare Dis 2011;6:3.ArticlePubMedPMC
- 112. Manto M, Honnorat J, Hampe CS, Guerra-Narbona R, López-Ramos JC, Delgado-García JM, et al. Disease-specific monoclonal antibodies targeting glutamate decarboxylase impair GABAergic neurotransmission and affect motor learning and behavioral functions. Front Behav Neurosci 2015;9:78.ArticlePubMedPMC
- 113. Ishida K, Mitoma H, Mizusawa H. Reversibility of cerebellar GABAergic synapse impairment induced by anti-glutamic acid decarboxylase autoantibodies. J Neurol Sci 2008;271:186–190.ArticlePubMed
- 114. Hampe CS, Petrosini L, De Bartolo P, Caporali P, Cutuli D, Laricchiuta D, et al. Monoclonal antibodies to 65kDa glutamate decarboxylase induce epitope specific effects on motor and cognitive functions in rats. Orphanet J Rare Dis 2013;8:82.ArticlePubMedPMC
- 115. Mitoma H, Ishida K, Shizuka-Ikeda M, Mizusawa H. Dual impairment of GABAA- and GABAB-receptor-mediated synaptic responses by autoantibodies to glutamic acid decarboxylase. J Neurol Sci 2003;208:51–56.ArticlePubMed
- 116. Ishida K, Mitoma H, Wada Y, Oka T, Shibahara J, Saito Y, et al. Selective loss of Purkinje cells in a patient with anti-glutamic acid decarboxylase antibody-associated cerebellar ataxia. J Neurol Neurosurg Psychiatry 2007;78:190–192.ArticlePubMed
- 117. Hadjivassiliou M. Advances in therapies of cerebellar disorders: immune-mediated ataxias. CNS Neurol Disord Drug Targets 2019;18:423–431.ArticlePubMed
- 118. Mitoma H, Manto M, Hampe CS. Immune-mediated cerebellar ataxias: practical guidelines and therapeutic challenges. Curr Neuropharmacol 2019;17:33–58.ArticlePubMedPMC
- 119. Takeguchi M, Nanri K, Okita M, Taguchi T, Ishiko T, Saitoh H. Efficacy of intravenous immunoglobulin for slowly progressive cerebellar atrophy. Rinsho Shinkeigaku 2006;46:467–474.PubMed
- 120. Fisher M. An unusual variant of acute idiopathic polyneuritis (syndrome of ophthalmoplegia, ataxia and areflexia). N Engl J Med 1956;255:57–65.ArticlePubMed
- 121. Mori M, Kuwabara S, Fukutake T, Yuki N, Hattori T. Clinical features and prognosis of Miller Fisher syndrome. Neurology 2001;56:1104–1106.ArticlePubMed
- 122. Hahn AF. Guillain-Barré syndrome. Lancet 1998;352:635–641.ArticlePubMed
- 123. Urushitani M, Udaka F, Kameyama M. Miller Fisher-Guillain-Barré overlap syndrome with enhancing lesions in the spinocerebellar tracts. J Neurol Neurosurg Psychiatry 1995;58:241–243.ArticlePubMedPMC
- 124. Kim YK, Kim JS, Jeong SH, Park KS, Kim SE, Park SH. Cerebral glucose metabolism in Fisher syndrome. J Neurol Neurosurg Psychiatry 2009;80:512–517.ArticlePubMed
- 125. Sandler RD, Hoggard N, Hadjivassiliou M. Miller-Fisher syndrome: is the ataxia central or peripheral? Cerebellum Ataxias 2015;2:3.ArticlePubMedPMC
- 126. Kornberg AJ, Pestronk A, Blume GM, Lopate G, Yue J, Hahn A. Selective staining of the cerebellar molecular layer by serum IgG in MillerFisher and related syndromes. Neurology 1996;47:1317–1320.ArticlePubMed
- 127. Berlit P, Rakicky J. The Miller Fisher syndrome. Review of the literature. J Clin Neuroophthalmol 1992;12:57–63.PubMed
- 128. Boronat A, Gelfand JM, Gresa-Arribas N, Jeong HY, Walsh M, Roberts K, et al. Encephalitis and antibodies to dipeptidyl-peptidase-like protein-6, a subunit of Kv4.2 potassium channels. Ann Neurol 2013;73:120–128.ArticlePubMed
- 129. Balint B, Jarius S, Nagel S, Haberkorn U, Probst C, Blöcker IM, et al. Progressive encephalomyelitis with rigidity and myoclonus: a new variant with DPPX antibodies. Neurology 2014;82:1521–1528.ArticlePubMed
- 130. Tobin WO, Lennon VA, Komorowski L, Probst C, Clardy SL, Aksamit AJ, et al. DPPX potassium channel antibody: frequency, clinical accompaniments, and outcomes in 20 patients. Neurology 2014;83:1797–1803.ArticlePubMedPMC
- 131. van Sonderen A, Ariño H, Petit-Pedrol M, Leypoldt F, Körtvélyessy P, Wandinger KP, et al. The clinical spectrum of Caspr2 antibody-associated disease. Neurology 2016;87:521–528.ArticlePubMedPMC
- 132. Joubert B, Gobert F, Thomas L, Saint-Martin M, Desestret V, Convers P, et al. Autoimmune episodic ataxia in patients with anti-CASPR2 antibody-associated encephalitis. Neurol Neuroimmunol Neuroinflamm 2017;4:e371.ArticlePubMedPMC
- 133. DeBellard ME, Tang S, Mukhopadhyay G, Shen YJ, Filbin MT. Myelin-associated glycoprotein inhibits axonal regeneration from a variety of neurons via interaction with a sialoglycoprotein. Mol Cell Neurosci 1996;7:89–101.ArticlePubMed
- 134. Zis P, Rao DG, Hoggard N, Sarrigiannis PG, Hadjivassiliou M. Anti-MAG associated cerebellar ataxia and response to rituximab. J Neurol 2018;265:115–118.ArticlePubMed
- 135. Pittock SJ, Debruyne J, Krecke KN, Giannini C, van den Ameele J, De Herdt V, et al. Chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids (CLIPPERS). Brain 2010;133:2626–2634.ArticlePubMed
- 136. Dudesek A, Rimmele F, Tesar S, Kolbaske S, Rommer PS, Benecke R, et al. CLIPPERS: chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids. Review of an increasingly recognized entity within the spectrum of inflammatory central nervous system disorders. Clin Exp Immunol 2014;175:385–396.ArticlePubMedPMC
- 137. Mitoma H, Manto M, Hampe CS. Immune-mediated cerebellar ataxias: from bench to bedside. Cerebellum Ataxias 2017;4:16.ArticlePubMedPMC
REFERENCES
Figure & Data
References
Citations
- Different Purkinje cell pathologies cause specific patterns of progressive gait ataxia in mice
Dick Jaarsma, Maria B. Birkisdóttir, Randy van Vossen, Demi W.G.D. Oomen, Oussama Akhiyat, Wilbert P. Vermeij, Sebastiaan K.E. Koekkoek, Chris I. De Zeeuw, Laurens W.J. Bosman
Neurobiology of Disease.2024; 192: 106422. CrossRef - Paraneoplastic Cerebellar Degeneration Associated with Breast Cancer: A Case Report and a Narrative Review
Rosario Luca Norrito, Maria Grazia Puleo, Chiara Pintus, Maria Grazia Basso, Giuliana Rizzo, Tiziana Di Chiara, Domenico Di Raimondo, Gaspare Parrinello, Antonino Tuttolomondo
Brain Sciences.2024; 14(2): 176. CrossRef - Immune‐mediated spastic ataxia masquerading as clinically probable multisystem atrophy in an elderly woman
Rithvik Ramesh, Anuhya Chadalawada, Pedapati Radhakrishna, Lakshmi Narasimhan Ranganathan, Philo Hazeena, Sundar Shanmugam, Deepa Avadhani
Clinical and Experimental Neuroimmunology.2024;[Epub] CrossRef - Paraneoplastic neurological syndromes: upgraded approaches to diagnosis
V. N. Grigoryeva, E. A. Ruina
Russian neurological journal.2024; 29(1): 4. CrossRef - Neuronal antibodies in nonparaneoplastic autoimmune cerebellar ataxias
Albert Saiz, Francesc Graus
Current Opinion in Neurology.2024;[Epub] CrossRef - Paraneoplastic Cerebellar Ataxia: A Case Report and Literature Review
静 刘
Advances in Clinical Medicine.2024; 14(03): 350. CrossRef - Autoimmunologische Kleinhirnerkrankungens
Niklas Vogel, Christian Hartmann, Sven Meuth, Nico Melzer
Nervenheilkunde.2023; 42(01/02): 73. CrossRef - Evaluation and management of acute high-grade immunotherapy-related neurotoxicity
Marcelo Sandoval, Adriana H. Wechsler, Zahra Alhajji, Jayne Viets-Upchurch, Patricia Brock, Demis N. Lipe, Aisha Al-breiki, Sai-Ching J. Yeung
Heliyon.2023; 9(3): e13725. CrossRef - Consensus Paper: Latent Autoimmune Cerebellar Ataxia (LACA)
Mario Manto, Marios Hadjivassiliou, José Fidel Baizabal-Carvallo, Christiane S Hampe, Jerome Honnorat, Bastien Joubert, Hiroshi Mitoma, Sergio Muñiz-Castrillo, Aasef G. Shaikh, Alberto Vogrig
The Cerebellum.2023; 23(2): 838. CrossRef - Proinflammatory activation of microglia in the cerebellum hyperexcites Purkinje cells to trigger ataxia
Shu-Tao Xie, Wen-Chu Fan, Xian-Sen Zhao, Xiao-Yang Ma, Ze-Lin Li, Yan-Ran Zhao, Fa Yang, Ying Shi, Hui Rong, Zhi-San Cui, Jun-Yi Chen, Hong-Zhao Li, Chao Yan, Qipeng Zhang, Jian-Jun Wang, Xiao-Yang Zhang, Xiao-Ping Gu, Zheng-Liang Ma, Jing-Ning Zhu
Pharmacological Research.2023; 191: 106773. CrossRef - Immune-related adverse events and immune checkpoint inhibitors: a focus on neurotoxicity and clinical management
Rosanna Ruggiero, Raffaella Di Napoli, Nunzia Balzano, Donatella Ruggiero, Consiglia Riccardi, Antonietta Anatriello, Andrea Cantone, Liberata Sportiello, Francesco Rossi, Annalisa Capuano
Expert Review of Clinical Pharmacology.2023; 16(5): 423. CrossRef - Immune-mediated ataxias: Guide to clinicians
Alex T. Meira, Marianna P.M. de Moraes, Matheus G. Ferreira, Gustavo L. Franklin, Flávio M. Rezende Filho, Hélio A.G. Teive, Orlando G.P. Barsottini, José Luiz Pedroso
Parkinsonism & Related Disorders.2023; 117: 105861. CrossRef - Gluten Ataxia: an Underdiagnosed Condition
Marios Hadjivassiliou, R. A. Grϋnewald
The Cerebellum.2022; 21(4): 620. CrossRef - Clinical Problem Solving: Decreased Level of Consciousness and Unexplained Hydrocephalus
Naomi Niznick, Ronda Lun, Daniel A. Lelli, Tadeu A. Fantaneanu
The Neurohospitalist.2022; 12(2): 312. CrossRef - Pharmacotherapy of cerebellar and vestibular disorders
João Lemos, Mario Manto
Current Opinion in Neurology.2022; 35(1): 118. CrossRef - Advances in the Pathogenesis of Auto-antibody-Induced Cerebellar Synaptopathies
Hiroshi Mitoma, Mario Manto
The Cerebellum.2022; 22(1): 129. CrossRef - A Breakdown of Immune Tolerance in the Cerebellum
Christiane S. Hampe, Hiroshi Mitoma
Brain Sciences.2022; 12(3): 328. CrossRef - Acute Cerebellar Inflammation and Related Ataxia: Mechanisms and Pathophysiology
Md. Sorwer Alam Parvez, Gen Ohtsuki
Brain Sciences.2022; 12(3): 367. CrossRef - A Case Report of Anti-PCA-2-Positive Autoimmune Cerebellitis
霞 董
Advances in Clinical Medicine.2022; 12(04): 3272. CrossRef - Cell-Autonomous Processes That Impair Xenograft Survival into the Cerebellum
Lorenzo Magrassi, Giulia Nato, Domenico Delia, Annalisa Buffo
The Cerebellum.2022; 21(5): 821. CrossRef - Diagnosis and Clinical Features in Autoimmune-Mediated Movement Disorders
Pei-Chen Hsieh, Yih-Ru Wu
Journal of Movement Disorders.2022; 15(2): 95. CrossRef - Autoimmune cerebellar ataxia associated with anti-leucine-rich glioma-inactivated protein 1 antibodies: Two pediatric cases
Zhang Weihua, Ren Haitao, Deng Jie, Ren Changhong, Zhou Ji, Zhou Anna, Guan Hongzhi, Ren Xiaotun
Journal of Neuroimmunology.2022; 370: 577918. CrossRef - Anti-dipeptidyl-peptidase-like protein 6 encephalitis with pure cerebellar ataxia: a case report
Jing Lin, Min Zhu, Xiaocheng Mao, Zeqing Jin, Meihong Zhou, Daojun Hong
BMC Neurology.2022;[Epub] CrossRef - Central Positional Nystagmus
Ana Inês Martins, André Jorge, João Lemos
Current Treatment Options in Neurology.2022; 24(10): 453. CrossRef - Paraneoplastic Ataxia: Antibodies at the Forefront Have Become Routine Biomarkers
Lazaros C. Triarhou, Mario Manto
The Cerebellum.2022; 22(4): 534. CrossRef - Rare Etiologies in Immune-Mediated Cerebellar Ataxias: Diagnostic Challenges
Marios Hadjivassiliou, Mario Manto, Hiroshi Mitoma
Brain Sciences.2022; 12(9): 1165. CrossRef - Paraneoplastic syndromes in neuro-ophthalmology
SimonJ Hickman
Annals of Indian Academy of Neurology.2022; 25(8): 101. CrossRef - Immunotherapies for the Effective Treatment of Primary Autoimmune Cerebellar Ataxia: a Case Series
Jiao Li, Bo Deng, Wenli Song, Keru Li, Jingwen Ai, Xiaoni Liu, Haocheng Zhang, Yi Zhang, Ke Lin, Guofu Shao, Chunfeng Liu, Wenhong Zhang, Xiangjun Chen, Yanlin Zhang
The Cerebellum.2022; 22(6): 1216. CrossRef - Evaluation and Management of Acute High-Grade Immunotherapy-Related Neurotoxicity
Marcelo Sandoval, Adriana H. Wechsler, Zahra Alhajji, Jayne Viets-Upchurch, Patricia A. Brock, Demis N. Lipe, Aisha Al-Buraiki, Sai-Ching Jim Yeung
SSRN Electronic Journal .2022;[Epub] CrossRef - Stiff-Eye Syndrome—Anti-GAD Ataxia Presenting with Isolated Ophthalmoplegia: A Case Report
Abel Dantas Belém, Thaís de Maria Frota Vasconcelos, Rafael César dos Anjos de Paula, Francisco Bruno Santana da Costa, Pedro Gustavo Barros Rodrigues, Isabelle de Sousa Pereira, Paulo Roberto de Arruda Tavares, Gabriela Studart Galdino, Daniel Aguiar Dia
Brain Sciences.2021; 11(7): 932. CrossRef - Update on Paraneoplastic Cerebellar Degeneration
Philipp Alexander Loehrer, Lara Zieger, Ole J. Simon
Brain Sciences.2021; 11(11): 1414. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite