E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 5(2); 2012 > Article
-
Original Article
Clinical Features and Disability Milestones in Multiple System Atrophy and Progressive Supranuclear Palsy - Sang-Wook Lee, Seong-Beom Koh
-
Journal of Movement Disorders 2012;5(2):42-47.
DOI: https://doi.org/10.14802/jmd.12010
Published online: October 30, 2012
Department of Neurology, Korea University College of Medicine, Seoul, Korea
- Corresponding author: Seong-Beom Koh, MD, PhD Department of Neurology, Korea University College of Medicine, 148 Gurodong-ro, Guro-gu, Seoul 152-703, Korea, Tel +82-2-2626-1250, Fax +82-2-2626-1257, E-mail parkinson@korea.ac.kr
- The authors have no financial conflicts of interest.
Copyright © 2012 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 23,623 Views
- 135 Download
- 12 Crossref
ABSTRACT
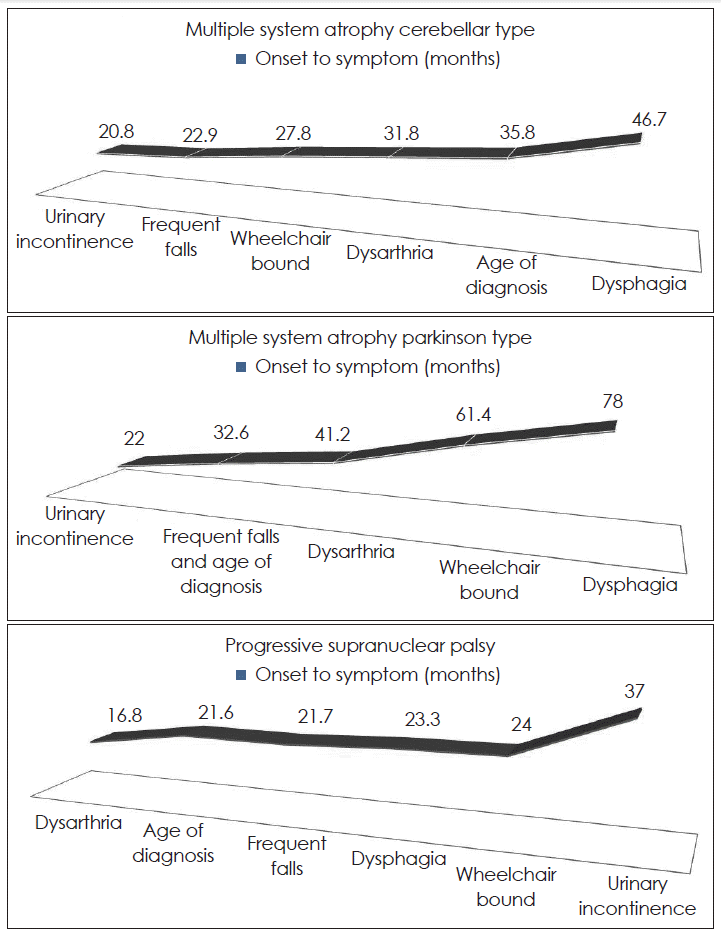
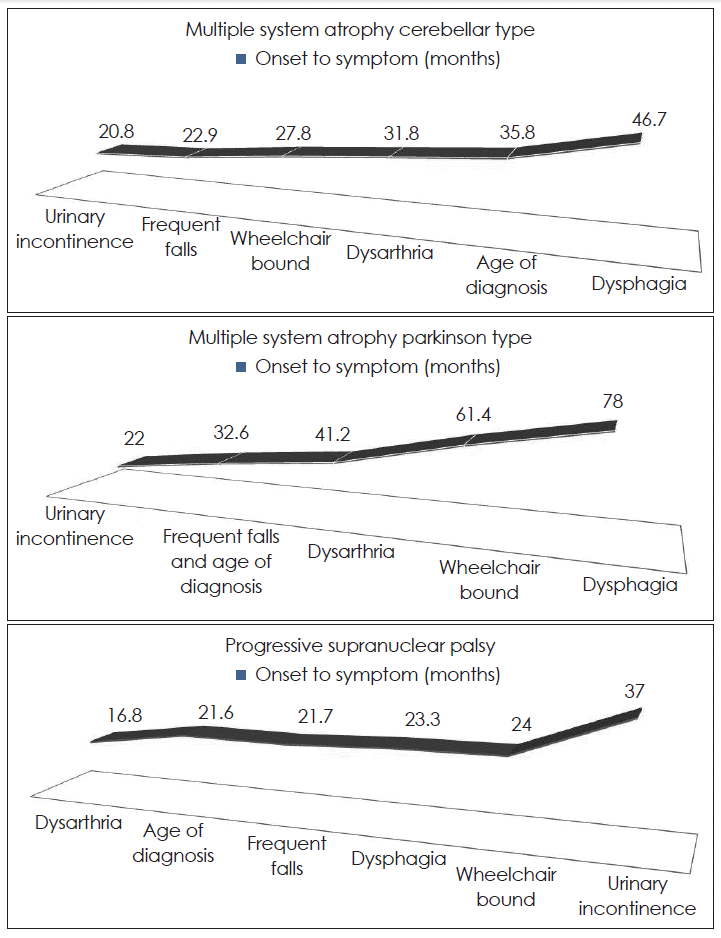
- Multiple system atrophy (MSA) and progressive supranuclear palsy (PSP) are an adult-onset progressive neurodegenerative disorder that are known to display diverse clinical features and disease progression. We aim to characterize the clinical features and disease progression in patients with MSA and PSP by using a number of relevant disability milestones in Koreans. Forty-one patients with MSA and 14 patients with PSP had been enrolled. The mean age at onset of MSA-C, MSA-P and PSP was 56.7 ± 7.8, 62.5 ± 8.0, 68.9 ± 6.1 years respectively. The most commonly reported symptom at disease onset is disequilibrium/dizziness in MSA-C, tremor in MSA-P and frequent falling in PSP. The mean duration of reaching milestones after disease onset in MSA-C were as followings: 20.8 (urinary incontinence), 22.9 (frequent falling), 27.8 (wheelchair bound), 31.8 (dysarthria) and 35.8 months (diagnosis). The mean duration of reaching milestones after disease onset were 22.0 (urinary incontinence), 32.6 (frequent falling and diagnosis), 41.2 (dysarthria), 61.4 months (wheelchair bound) in MSA-P and 16.8 (dysarthria), 21.6 (diagnosis), 21.7 (frequent falling), 24.0 months (wheel chair bound) in PSP. In the case of MSA, dizziness may occur for the first time. Thus, when the patient complains of non-specific dizziness, a follow-up examination to distinguish it from MSA can be helpful. There was a trend for patients with MSA-C to reach more disability milestones than in MSA-P and PSP before diagnosis. It may explain why patients with MSA-C are required more detail history taking and neurologic examination at an earlier stage.
- Multiple system atrophy (MSA) is a sporadic, progressive, adult-onset disease characterized by parkinsonism, cerebellar syndrome and autonomic dysfunction.1 Mean survival ranges between seven and nine years after initial clinical presentation. However, the early development of autonomic dysfunction, female gender, older age of onset, a short interval from disease onset to reaching the first clinical milestone and not being admitted to residential care were predictive factors for rapid disease progression and shorter survival in patients with MSA.2,3
- Progressive supranuclear palsy (PSP) is an adult-onset neurodegenerative disorder characterized by early postural instability, which leads to falls, and a vertical supranuclear-gaze palsy.4 Unlike typical Parkinson’s disease, falls begin within the first year and by year 3 they are common unless precautions are taken to prevent them. The assault on balance virtually warrants that most patients will end up wheelchair bound within 4 to 7 years.5 And time from disease onset to the development of other clinical features depended on the clinical feature. On average, falls were the earliest feature (median latency 0 years, range 0 to 16), speech problems (1.75, range 0 to 15), swallowing problems (3.58, range 0 to 16), and insertion of a PEG system (5.0, range 0.58 to 17 years).6
- However, it is based on a survey conducted in Europe. Fifty MSA patients were recruited consecutively at eight European MSA Study Group centers. Twenty nine patients (58%) showed a predominance of parkinsonian signs (MSA-P), and 42% of them showed a predominance of cerebellar dysfunction (MSA-C).7 This is similar to the results of 67 MSA patients at nine United States movement disorder centers.8
- In Japan, four clinics were participated in analyzing 142 MSA cases. A total of 119 patients (83.8%) were classified as having MSA-C and 23 patients (16.2%) were classified as having MSA-P.9 It is different results conducted in Europe and United States.
- In addition, the average of the onset of symptoms is known as early 70 in Japanese PSP patients. It is different from European data that the average of the onset of symptoms is known as mid 60.5,10,11
- This shows that the disease progression may vary depending on environmental factors or ethnic differences.1,9,12,13 In South Korea, it makes difficult to accept the clinical course based on a result of research conducted in Europe and United States. Nevertheless, the current demographic statistics in Korea has not been established.
- Thus, we aim to characterize the clinical symptoms and progress of symptoms over time in patients with MSA and PSP by using a number of relevant disability milestones in Koreans. It could be paid enough attention to the patients before developing disabilities and if a failure occurs due to each disease it could enable an appropriate management based on this survey.
Introduction
- Patients
- Data for this analysis were obtained from the electronic database of the Korea University Guro Hospital Parkinson center from 2010 to 2011. Forty-one patients with MSA and 14 patients with PSP had been enrolled. The diagnosis of MSA was made according to established criteria and the diagnosis of PSP was made according to the National Institute for Neurological Diseases and Stroke-Society for PSP (NINDS-SPSP) criteria.14,15 All patients consented to the data analyses. This study was approved by the Institutional Review Board at the Korea University Guro Hospital.
- Medical record review
- We retrospectively reviewed medical records to investigate the history of MSA and PSP. And we carried out neurological examination and the interviews for outpatients to assess the degree of disability depending on the progression of the disease. Motor feature (frequent falling, bradykinesia, tremor), autonomic feature (urinary incontinence), dysarthria, dysphagia and wheelchair bound were determined.
- Five milestones of disease progression were selected on the basis that all were likely to require additional medical attention, and to be well documented in the medical records. These were: urinary incontinence, frequent falling (defined as falls occurring more than twice per year, or the documentation of ‘frequent’ or ‘regular’ falls),2 dependence on wheelchair for mobility, dysarthria and dysphagia.
- The disease onset is defined by the time of initial manifestation of any motor or autonomic feature: Parkinsonism, disequilibrium, frequent falling, gait difficulty, dizziness, dysarthria, dysphagia, urinary frequency, residual urine and urinary incontinence. Symptoms were recorded as being absent if they were not mentioned in the notes. And we noted the time that the symptom was first mentioned in the medical records.
- In most cases the year of onset was available. But where the month of onset of each milestone was not noted, July was used as the midpoint of the year for analysis.
- Statistical analysis
- Chi-square was used to compare categorical variables and one-way ANOVA, Mann-Whitney U-test or Kruskal-Wallis test, as appropriate, was used to compare continuous variables between subgroups. Statistical analyses of data were performed with SPSS version 12.0.
Methods
- Demographic and Clinical background informations
- The demographic data and parkinsonian features were compared between cases with MSA and PSP in Table 1. We identified 41 cases of MSA and 14 cases of PSP for review. Of the 41 cases of MSA, 20 (48.8%) had MSA–C and 21 (51.2%) had MSA-P. The overall gender distribution (male/female) was 1.5 : 1, while it was 1 : 1 in MSA-C, 1.3 : 1 in MSA-P and 3.7 : 1 in PSP. The mean follow-up period of MSA patients was 2.4 years. And the mean follow-up period of PSP patients was 2.0 years.
- The mean age at onset of MSA-C, MSA-P and PSP was 56.7 ± 7.8, 62.5 ± 8.0, 68.9 ± 6.1 years, respectively (p valuea: < 0.001, p valueb: 0.043, p valuec: 0.045, Comparison of aMSA-C and PSP, bMSA-P and PSP, cMSA-C and MSA-P). And the mean age at diagnosis of MSA-C, MSA-P and PSP was 59.7 ± 7.4, 65.2 ± 8.1, 70.7 ± 6.3 years respectively (p valuea: < 0.001, p valueb: 0.096, p valuec: 0.055, Comparison of aMSA-C and PSP, bMSA-P and PSP, cMSA-C and MSA-P). Age at onset and diagnosis were older in PSP than in MSA-C.
- Disequilibrium is most frequent initial sign of MSA-C (50%). Since then, followed by dizziness (15%), dysarthria (15%) and urinary symptoms (15%). Tremor is most frequent initial sign of MSA-P (24%). But after that, the distribution of initial sign in MSA-P vary: Disequilibrium (19%), dysarthria (14%), bradykinesia (10%), dizziness (10%), frequent falling (10%) and urinary frequency (10%). Frequent falling is most frequent initial sign of PSP (43%). Since then, followed by tremor (29%), bradykinesia (14%) and gait difficulty (14%).
- One patient with MSA-P died in our study. The cause of death was pneumonia and the time from the onset of disease to death was 7.9 years.
- Clinical milestones
- Dysarthria was the most common milestone in MSA-C (80%). Urinary incontinence was the most common milestone in MSA-P (61.9%) and frequent falls was the most common milestone in PSP (71.4%)(Table 2). In MSA-C and MSA-P, the second frequent milestone was frequent falls (65%, 47.6% respectively) whereas in PSP it was dysphagia (42.9%). There were significant differences in the frequency of urinary incontinence (p = 0.001), wheelchair bound (p = 0.011), dysarthria (p = 0.004) and dysphagia (p < 0.001) between subgroups. However, there was no differences in duration of clinical milestones between subgroups (Table 3). Urinary incontinence was the first milestone in MSA and dysarthria was the first milestone in PSP (Figure 1).
Results
- In the multicentre registry of the German Competence Network on Parkinson’s disease, information for 221 patients with probable multiple system atrophy is as follows: age at onset 60 ± 9 yrs (range: 34–83 yrs) and the male = female ratio was 0.9 : 1.16 This is similar to the results of 67 MSA patients at nine United States movement disorder centers. Mean age at MSA onset was 60.5 ± 9.9 years. Most patients had MSA-P (60%) and the male = female ratio was 1.5 : 1.8 Other previous studies investigating the clinical characteristics of MSA have identified similar features (Table 4).2,12,17–21
- Forty-one probable MSA patients were recruited at our clinics. 51.2% of patients showed a predominance of parkinsonian signs (MSA-P), and 48.8% of them showed a predominance of cerebellar dysfunction (MSA-C). Age at onset 59.7 ± 8.3 yrs, and the male = female ratio was 1.2 : 1.
- However, MSA-C is considered to be the more common subtypes of MSA in the Spanish and Japanese population.9,13,20 It is different from other studies. Köllensperger et al.20 suggest the high proportion of MSA-C patients in Spain is due to the expertise in ataxias of one of the two participating centres from this country.
- The difference of disease manifestations between countries suggests that environmental factors and genetic factors may influence the clinical phenotype of MSA.13,18,22 However, considering that Korea is environmentally and genetically close to Japan, the predominance of MSA-C in Japanese population may be caused by another factor.
- Yabe et al.9 suggest a change of clinical manifestations was observed in some of MSA-C patients, a propotion of MSA subtypes can vary by the time of long-term follow-up. Therefore, the propotion of MSA subtypes can vary depending on how long period of time passed from the disease onset at the time of investigation. Because of the difficulty in diagnosis of MSA can also lead to a difference in the ratio between MSA subgroups.
- In PSP, male predominance was consistent with previously reported studies.2,10,23–29 But the age of onset of the PSP in Korea is older than other ethnics (Table 5). Genetic studies have detected an association between the presence of the tau gene A0 allele and patients with PSP. The detection of significantly lower age at onset with the A0/A0 alleles is consistent with the known association of this genotype as a risk factor for PSP.21 So it could be a significant overrepresentation of the tau A0 allele in Korean patients with PSP is less frequent. However, according to the assumptions that Japanese and Koreans have similar genetic background, tau gene A0 allele also could be more frequent in Korean patients. It may show this repeat may not be causal for PSP but represents a marker for other molecular genetic risk factors within or close to the tau gene on chromosome 17 than the role of this dinucleotide repeat in PSP may be different between ethnics.30
- It is also consistent with previously reported studies that age at onset was significantly older in PSP than in the MSA sub-types.2,31
- Dysarthria was the most common milestone in MSA-C (80%). Urinary incontinence was the most common milestone in MSA-P (61.9%) and frequent falls was the most common milestone in PSP (71.4%)(Table 3). In MSA-C and MSA-P, the second frequent milestone was frequent falls (65%, 47.6% respectively) whereas in PSP it was dysphagia (42.9%). O’Sullivan et al.2 suggest that urinary catheter was the most common milestone in MSA-C (71%), frequent falls in MSA-P and PSP (64%, 82% respectively) followed by wheelchair bound (55%, 56%, 46% respectively). Frequent falls are relatively frequent milestones in MSA and PSP than other clinical milestones. Wheelchair bound is less frequent milestones than O’Sullivan et al.’s study. It might be that we did not have pathological confirmation by autopsy because only one patient deceased and mean follow-up period after disease onset is just 5 years.
- The MSA tends to be diagnosed when there is urinary difficulty and the PSP is diagnosed when there is frequent falling. In the case of MSA, we assumed disequilibrium and dizziness as the first manifestation of MSA when patients with MSA referred to our clinics initially, but they didn’t come to our clinics before urinary difficulty was developed. It is probably because the above symptoms haven’t significant impact on their lives and they regarded disequilibrium and dizziness as just senile change.
- This study has several limitations. First, there may have been some bias because the onset time of symptoms set through a retrospective study. Second, because the sensitivity of the clinical diagnostic criteria for MSA without pathological findings has been relatively low.32 Therefore, there could be a patient without MSA who was diagnosed with MSA currently. Finally, neuropathological examination is also the “gold standard” for diagnosis of PSP. According to a study, only three patients (42.9%) matched the clinical diagnostic criteria of PSP proposed by the NINDS-SPSP at the time of death. In addition, only one patient (14.3%) matched these criteria at the time of the initial symptoms. Such underdiagnosis of PSP was mainly caused by heterogeneity, variety of the timing, and presence of symptoms in exclusion criteria.10 So pathological confirmation is required for accurate diagnosis in MSA and PSP.
- This study was designed to characterize the clinical symptoms and progress of symptoms over time in patients with MSA and PSP by using a number of relevant disability milestones in Koreans.
- Until now, the onset of MSA was defined as the time when initially noted Parkinsonism or autonomic symptoms. However, in the case of MSA, dizziness may occur for the first time. Thus, when the patient complains of not-specific dizziness, a follow-up examination to distinguish it from MSA can be helpful.
- There was a trend for patients with MSA-C to reach more disability milestones than in MSA-P and PSP before diagnosis. It may explain why patients with MSA-C required more detail history taking and neurologic examination at an earlier stage.
Discussion
| MSA-C | MSA-P | PSP | p valuea | p valueb | p valuec | Chi-square | |
|---|---|---|---|---|---|---|---|
| Total number | 20 | 21 | 14 | ||||
| Male/Female | 10/10 | 12/9 | 11/3 | 0.040* | |||
| Age of onset (years) | 56.7 ± 7.8 | 62.5 ± 8.0 | 68.9 ± 6.1 | < 0.001* | 0.043* | 0.045* | |
| Age of diagnosis (years) | 59.7 ± 7.4 | 65.2 ± 8.1 | 70.7 ± 6.3 | < 0.001* | 0.096 | 0.055 |
Chi-square was used to compare categorical variables and one-way ANOVA was used to compare continuous variables between subgroups.
a Comparison of MSA-C and PSP,
b Comparison of MSA-P and PSP,
c Comparison of MSA-C and MSA-P.
* significant p value < 0.05. MSA: multiple system atrophy, PSP: progressive supranuclear palsy.
| MSA-C (n = 20) | MSA-P (n = 21) | PSP (n = 14) | Chi-square | |
|---|---|---|---|---|
| Urinary incontinence | 9 (45) | 13 (61.9) | 3 (21) | 0.001* |
| Frequent falling | 13 (65) | 10 (47.6) | 10 (71.4) | 0.075 |
| Wheelchair bound | 4 (20) | 7 (33.3) | 1 (7) | 0.011* |
| Dysarthria | 16 (80) | 9 (42.9) | 5 (36) | 0.004* |
| Dysphagia | 7 (35) | 1 (4.8) | 6 (42.9) | < 0.001* |
| MSA-C | MSA-P | PSP | p valuea | p valueb | p valuec | |
|---|---|---|---|---|---|---|
| Urinary incontinence | 20.8 | 22.0 | 37.0 | 0.282 | 0.439 | 0.794 |
| Frequent falling | 22.9 | 32.6 | 21.7 | 0.693 | 0.436 | 0.784 |
| Wheelchair bound | 27.8 | 61.4 | 24.0 | 0.800 | 0.750 | 0.527 |
| Dysarthria | 31.8 | 41.2 | 16.8 | 0.968 | 0.518 | 0.487 |
| Dysphagia | 46.7 | 78.0 | 23.3 | 0.366 | 0.286 | 0.500 |
| Onset to diagnosis | 35.8 | 32.6 | 21.6 | 0.545 | 0.263 | 0.794 |
Kruskal-Wallis test and Mann-Whitney U-test were used to assess differences of duration between MSA subgroups and PSP. Duration of clinical milestones are expressed as months.
a Comparison of MSA-C and PSP,
b Comparison of MSA-P and PSP,
c Comparison of MSA-C and MSA-P.
* significant p value < 0.017 (= 0.05/3). MSA: multiple system atrophy, PSP: progressive supranuclear palsy.
| Study (n) | Age at onset (years) | Male : Female | MSA-P (%) |
|---|---|---|---|
| Queen Square Brain Bank for Neurological Disorders (83)2 | 56.8 ± 10.2 | 37:46 | NA |
| Nine US Movement Disorder Centers (67)8 | 60.5 ± 9.9 | 40:27 | 60 |
| Neurology Clinics of Hokkaido University Hospital, Hokuyukai Neurology Hospital, Sapporo Minami National Hospital, or Ebetsu City Hospital (142)9 | 58.2 ± 7.1 | 84:58 | 16.20 |
| German Competence Network on Parkinson’s disease (221)16 | 60 ± 9 | NA | 84 |
| Institute of Neurology (100)17 | 53 | 67:33 | 82 |
| Network of 120 Public and Private Practitioners in Gironde (50)18 | 62.1 | 27:23 | 70 |
| Movement Disorders Clinic, Tan Iock Seng Hospital (33)19 | 60 ± 10 | 20:13 | 33 |
| 19 European MSA Study Group centres (437)20 | 57.8 | 223:214 | 68.2 |
| Seoul National University Hospital Movement Disorder Clinic (455)12 | 60.18 ± 8.8 | 222:233 | 54.95 |
| Rabat University Hospital (17)21 | 52 ± 9 | 13:4 | 82.40 |
| Korea University Guro Hospital Parkinson center (41) | 59.7 ± 8.3 | 22:19 | 52.50 |
| Study (n) | Age at onset (years) | Male : Female |
|---|---|---|
| Queen Square Brain Bank for Neurological Disorders (98)2 | 66.5 (RS), 63.2 (PSP-P) | 63 : 35 |
| Yokufukai Geriatric Hospital (14)10 | 72.1 ± 7.1 | 7 : 7 |
| Seven medical centres of four countries (Austria, England, France, and the United States) (24)23 | 63 | 15 : 9 |
| Movement Disorder Unit of the Department of Neurology of São Paulo University Medical School (16)24 | 64.75 ± 7.28 | 10 : 6 |
| Clinica Neurologica II, Policlinico, Piazza Giulio Cesare 11 (25)25 | 62 ± 6.2 | 15 : 10 |
| Centro Internacional de Restauracion Neurologica (18)26 | 58.6 ± 8.2 | 10 : 8 |
| Sara Koe PSP Research Centre (103)27 | 66.4 | 65 : 38 |
| University of Miami Brain Endowment Bank (22)28 | 67.6 ± 9.5 | 15 : 7 |
| Higashi Nagoya National Hospital (45)29 | 64.9 | 28 : 17 |
| Mayo Clinic (121)33 | 66.2 (Male), 68.5 (Female) | 53 : 68 |
| Korea University Guro Hospital Parkinson center (14) | 68.9 ± 6.1 | 11 : 3 |
- 1. Stefanova N, Bücke P, Duerr S, Wenning GK. Multiple system atrophy: an update. Lancet Neurol 2009;8:1172–1178.ArticlePubMed
- 2. O’Sullivan SS, Massey LA, Williams DR, Silveira-Moriyama L, Kempster PA, Holton JL, et al. Clinical outcomes of progressive supra-nuclear palsy and multiple system atrophy. Brain 2008;131(Pt 5):1362–1372.ArticlePubMed
- 3. Tada M, Onodera O, Tada M, Ozawa T, Piao YS, Kakita A, et al. Early development of autonomic dysfunction may predict poor prognosis in patients with multiple system atrophy. Arch Neurol 2007;64:256–260.ArticlePubMed
- 4. Burn DJ, Lees AJ. Progressive supranuclear palsy: where are we now? Lancet Neurol 2002;1:359–369.ArticlePubMed
- 5. Lubarsky M, Juncos JL. Progressive supranuclear palsy: a current review. Neurologist 2008;14:79–88.ArticlePubMed
- 6. Nath U, Ben-Shlomo Y, Thomson RG, Lees AJ, Burn DJ. Clinical features and natural history of progressive supranuclear palsy: a clinical cohort study. Neurology 2003;60:910–916.ArticlePubMed
- 7. Geser F, Wenning GK, Seppi K, Stampfer-Kountchev M, Scherfler C, Sawires M, et al. Progression of multiple system atrophy (MSA): a prospective natural history study by the European MSA Study Group (EMSA SG). Mov Disord 2006;21:179–186.ArticlePubMed
- 8. May S, Gilman S, Sowell BB, Thomas RG, Stern MB, Colcher A, et al. Potential outcome measures and trial design issues for multiple system atrophy. Mov Disord 2007;22:2371–2377.ArticlePubMed
- 9. Yabe I, Soma H, Takei A, Fujiki N, Yanagihara T, Sasaki H. MSA-C is the predominant clinical phenotype of MSA in Japan: analysis of 142 patients with probable MSA. J Neurol Sci 2006;249:115–121.ArticlePubMed
- 10. Sakamoto R, Tsuchiya K, Mimura M. Clinical heterogeneity in progressive supranuclear palsy: problems of clinical diagnostic criteria of NINDS-SPSP in a retrospective study of seven Japanese autopsy cases. Neuropathology 2010;30:24–35.ArticlePubMed
- 11. Kawashima M, Miyake M, Kusumi M, Adachi Y, Nakashima K. Prevalence of progressive supranuclear palsy in Yonago, Japan. Mov Disord 2004;19:1239–1240.ArticlePubMed
- 12. Kim HJ, Jeon BS, Lee JY, Yun JY. Survival of Korean patients with multiple system atrophy. Mov Disord 2011;26:909–912.ArticlePubMed
- 13. Watanabe H, Saito Y, Terao S, Ando T, Kachi T, Mukai E, et al. Progression and prognosis in multiple system atrophy: an analysis of 230 Japanese patients. Brain 2002;125(Pt 5):1070–1083.ArticlePubMed
- 14. Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDSSPSP international workshop. Neurology 1996;47:1–9.ArticlePubMed
- 15. Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71:670–676.ArticlePubMedPMC
- 16. Wüllner U, Schmitz-Hübsch T, Abele M, Antony G, Bauer P, Eggert K. Features of probable multiple system atrophy patients identified among 4770 patients with parkinsonism enrolled in the multicentre registry of the German Competence Network on Parkinson’s disease. J Neural Transm 2007;114:1161–1165.ArticlePubMed
- 17. Wenning GK, Ben Shlomo Y, Magalhães M, Daniel SE, Quinn NP. Clinical features and natural history of multiple system atrophy. An analysis of 100 cases. Brain 1994;117(Pt 4):835–845.ArticlePubMed
- 18. Chrysostome V, Tison F, Yekhlef F, Sourgen C, Baldi I, Dartigues JF. Epidemiology of multiple system atrophy: a prevalence and pilot risk factor study in Aquitaine, France. Neuroepidemiology 2004;23:201–218.ArticlePubMed
- 19. Jamora RD, Gupta A, Tan AK, Tan LC. Clinical characteristics of patients with multiple system atrophy in Singapore. Ann Acad Med Singapore 2005;34:553–557.PubMed
- 20. Köllensperger M, Geser F, Ndayisaba JP, Boesch S, Seppi K, Ostergaard K, et al. Presentation, diagnosis, and management of multiple system atrophy in Europe: final analysis of the European multiple system atrophy registry. Mov Disord 2010;25:2604–2612.ArticlePubMed
- 21. Regragui W, Lachhab L, Razine R, Benjelloun H, Ait Benhaddou EH, Benomar A, et al. Profile of multiple system atrophy in Moroccan patients attending a movement disorders outpatient clinic in Rabat university hospital. Rev Neurol (Paris). 2012. [Epub ahead of print]. Article
- 22. Bower JH, Maraganore DM, McDonnell SK, Rocca WA. Incidence of progressive supranuclear palsy and multiple system atrophy in Olmsted County, Minnesota, 1976 to 1990. Neurology 1997;49:1284–1288.ArticlePubMed
- 23. Litvan I, Mangone CA, McKee A, Verny M, Parsa A, Jellinger K, et al. Natural history of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) and clinical predictors of survival: a clinicopathological study. J Neurol Neurosurg Psychiatry 1996;60:615–620.ArticlePubMedPMC
- 24. Carrilho PE, Barbosa ER. Progressive supranuclear palsy in a sample of Brazilian population: clinical features of 16 patients. Arq Neuropsiquiatr 2002;60:917–922.ArticlePubMed
- 25. Diroma C, Dell’Aquila C, Fraddosio A, Lamberti S, Mastronardi R, Russo I, et al. Natural history and clinical features of progressive supranuclear palsy: a clinical study. Neurol Sci 2003;24:176–177.ArticlePubMed
- 26. Alvarez-González E, Maragoto-Rizo C, Arteche-Prior M, Pérez-Parra S, Carballo M, Alvarez-González L. [A clinical and epidemiological description of a series of patients diagnosed as suffering from progressive supranuclear palsy]. Rev Neurol 2004;39:1006–1010.PubMed
- 27. Williams DR, de Silva R, Paviour DC, Pittman A, Watt HC, Kilford L, et al. Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson’s syndrome and PSP-parkinsonism. Brain 2005;128(Pt 6):1247–1258.ArticlePubMed
- 28. Papapetropoulos S, Gonzalez J, Mash DC. Natural history of progressive supranuclear palsy: a clinicopathologic study from a population of brain donors. Eur Neurol 2005;54:1–9.ArticlePubMed
- 29. Aiba I, Saito Y, Tamakoshi A, Matsuoka Y. [Prognosis of patients with progressive supranuclear palsy]. Rinsho Shinkeigaku 2005;45:565–570.PubMed
- 30. Conrad C, Amano N, Andreadis A, Xia Y, Namekataf K, Oyama F, et al. Differences in a dinucleotide repeat polymorphism in the tau gene between Caucasian and Japanese populations: implication for progressive supranuclear palsy. Neurosci Lett 1998;250:135–137.ArticlePubMed
- 31. Testa D, Monza D, Ferrarini M, Soliveri P, Girotti F, Filippini G. Comparison of natural histories of progressive supranuclear palsy and multiple system atrophy. Neurol Sci 2001;22:247–251.ArticlePubMed
- 32. Osaki Y, Wenning GK, Daniel SE, Hughes A, Lees AJ, Mathias CJ, et al. Do published criteria improve clinical diagnostic accuracy in multiple system atrophy? Neurology 2002;59:1486–1491.ArticlePubMed
- 33. Baba Y, Putzke JD, Whaley NR, Wszolek ZK, Uitti RJ. Progressive supranuclear palsy: phenotypic sex differences in a clinical cohort. Mov Disord 2006;21:689–692.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
- Disease course and treatment patterns in progressive supranuclear palsy: A real-world study
John C. Morgan, Xiaolan Ye, Jennifer A. Mellor, Keisha J. Golden, Jorge Zamudio, Louis A. Chiodo, Yanjun Bao, Tao Xie
Journal of the Neurological Sciences.2021; 421: 117293. CrossRef - Progression of Oropharyngeal Dysphagia in Patients with Multiple System Atrophy
Hui Jae Do, Han Gil Seo, Hyun Haeng Lee, Byung-Mo Oh, Yoon Kim, Aryun Kim, Han-Joon Kim, Beomseok Jeon, Tai Ryoon Han
Dysphagia.2020; 35(1): 24. CrossRef - Is There a Difference in Autonomic Dysfunction Between Multiple System Atrophy Subtypes?
Divyani Garg, Achal Kumar Srivastava, Ashok Kumar Jaryal, Roopa Rajan, Akanksha Singh, Awadh Kishor Pandit, Deepti Vibha, Garima Shukla, Ajay Garg, Ravindra Mohan Pandey, Kameshwar Prasad
Movement Disorders Clinical Practice.2020; 7(4): 405. CrossRef - Models of multiple system atrophy
He-Jin Lee, Diadem Ricarte, Darlene Ortiz, Seung-Jae Lee
Experimental & Molecular Medicine.2019; 51(11): 1. CrossRef - Multiple System Atrophy: An Oligodendroglioneural Synucleinopathy1
Kurt A. Jellinger, George Perry, Jesus Avila, Massimo Tabaton, Xiongwei Zhu
Journal of Alzheimer's Disease.2018; 62(3): 1141. CrossRef - Present and future of disease-modifying therapies in multiple system atrophy
Miguel Lopez-Cuina, Alexandra Foubert-Samier, François Tison, Wassilios G. Meissner
Autonomic Neuroscience.2018; 211: 31. CrossRef - Different subregional metabolism patterns in patients with cerebellar ataxia by 18F-fluorodeoxyglucose positron emission tomography
Minyoung Oh, Jae Seung Kim, Jungsu S. Oh, Chong Sik Lee, Sun Ju Chung, Byeong-Cheol Ahn
PLOS ONE.2017; 12(3): e0173275. CrossRef - Vestibular Deficits in Neurodegenerative Disorders: Balance, Dizziness, and Spatial Disorientation
Thomas Cronin, Qadeer Arshad, Barry M. Seemungal
Frontiers in Neurology.2017;[Epub] CrossRef - Progressive supranuclear palsy: progression and survival
Julieta E. Arena, Stephen D. Weigand, Jennifer L. Whitwell, Anhar Hassan, Scott D. Eggers, Günter U. Höglinger, Irene Litvan, Keith A. Josephs
Journal of Neurology.2016; 263(2): 380. CrossRef - Multiple system atrophy: pathogenic mechanisms and biomarkers
Kurt A. Jellinger, Gregor K. Wenning
Journal of Neural Transmission.2016; 123(6): 555. CrossRef - Urinary Dysfunction in Progressive Supranuclear Palsy Compared with Other Parkinsonian Disorders
Tatsuya Yamamoto, Fuyuki Tateno, Ryuji Sakakibara, Shogo Furukawa, Masato Asahina, Tomoyuki Uchiyama, Shigeki Hirano, Yoshitaka Yamanaka, Miki Fuse, Yasuko Koga, Mitsuru Yanagisawa, Satoshi Kuwabara, Jong-Ling Fuh
PLOS ONE.2016; 11(2): e0149278. CrossRef - Movement and Other Neurodegenerative Syndromes in Patients with Systemic Rheumatic Diseases
Rikitha Menezes, Alexander Pantelyat, Izlem Izbudak, Julius Birnbaum
Medicine.2015; 94(31): e0971. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite