Cerebrospinal Fluid Amyloid β1-42, Tau, and Alpha-Synuclein Predict the Heterogeneous Progression of Cognitive Dysfunction in Parkinson’s Disease
Article information
Abstract
Parkinson’s disease (PD) is a neurodegenerative disease with heterogeneous pathological and clinical features. Cognitive dysfunction, a frequent non-motor complication, is a risk factor for poor prognosis and shows inter-individual variation in its progression. Of the clinical studies performed to identify biomarkers of PD progression, the Parkinson’s Progression Markers Initiative (PPMI) study is the largest study that enrolled drug-naïve and very early stage PD patients. The baseline characteristics of the PPMI cohort were recently published. The diagnostic utility of cerebrospinal fluid (CSF) biomarkers, including alpha-synuclein (α-syn), total tau, phosphorylated tau at Thr181, and amyloid β1-42, was not satisfactory. However, the baseline data on CSF biomarkers in the PPMI study suggested that the measurement of the CSF biomarkers enables the prediction of future cognitive decline in PD patients, which was consistent with previous studies. To prove the hypothesis that the interaction between Alzheimer’s pathology and α-syn pathology is important to the progression of cognitive dysfunction in PD, longitudinal observational studies must be followed. In this review, the neuropathological nature of heterogeneous cognitive decline in PD is briefly discussed, followed by a summarized interpretation of baseline CSF biomarkers derived from the data in the PPMI study. The combination of clinical, biochemical, genetic and imaging biomarkers of PD constitutes a feasible strategy to predict the heterogeneous progression of PD.
INTRODUCTION
Parkinson’s disease (PD) is a synucleinopathy with heterogeneous progression of motor and non-motor dysfunction. Several decades ago, motor phenotypes were suggested as classifiers of PD heterogeneity [1,2]. In fact, several well-designed studies provided evidence that PD patients with the postural instability and gait disturbance (PIGD) phenotype of motor symptoms showed a rapid progression of functional disability and cognitive deficit compared with patients with the tremor-dominant (TD) motor phenotype [1-5]. Although the mechanisms of the inherent heterogeneous progression of PD are still largely unknown, the heterogeneity of the disease may be associated with alterations of neural connectivity between the motor and cognitive control areas of the brain. The early development of dementia in PD patients is a major risk factor for poor prognosis and high mortality. Therefore, the early detection of patients who are likely to develop dementia rapidly is crucial. It has been established that the overall prevalence of cognitive dysfunction without functional deficit sufficient for the diagnosis of dementia (i.e., cognitive impairment) in PD is approximately 30% [6,7], and ~80% of patients with PD will develop overt dementia over 20 years after disease onset [8,9]. There are currently no proven underlying pathogenic mechanisms of cognitive decline in PD patients. Furthermore, several well-designed clinical studies to identify biochemical, imaging and genetic biomarkers of PD are emerging. Among the large multicenter longitudinal PD biomarker studies, the Parkinson Progression Marker Initiative (PPMI) is the largest study to homogeneously enroll drug-naïve PD patients at very early stages of the disease. The median duration of disease (from diagnosis to enrollment) was only 4.2 months (range, 0.03–38.83), and the Hoehn and Yahr stage (H&Y) of 99.5% of PPMI patients is I or II [10]. The primary objective of the PPMI study was to identify the biomarkers that predict PD progression [11]. This review briefly provides evidence of the heterogeneous progression in the cognitive decline of PD patients, discusses the pathophysiologic mechanisms related to heterogeneous cognitive decline, and summarizes recent advances in the development of cerebrospinal fluid (CSF) biomarkers to predict disease progression in the PPMI study.
EVIDENCE OF COGNITIVE HETEROGENEITY IN PD
Clinical characteristics associated with cognitive heterogeneity
The existence of subgroups within PD with distinct clinical patterns of motor and non-motor symptoms is widely accepted [12,13]. The clinical heterogeneity of PD may be associated with a variability in pathogenic mechanisms that may be controlled by inherited and environmental factors (Figure 1). Based on the motor symptoms of PD patients, formulas were developed to define the motor phenotypes using Movement Disorder Society Unified Parkinson’s Disease Rating Scale (MDS-UPDRS), a revision of the original UPDRS [14]. The motor phenotypes are most often classified by calculating the ratio between the scores of tremor-related and balance/gait/posture-related items in MDS-UPDRS [2]. Using this system, PD patients can be easily classified as having the TD or PIGD phenotype. However, despite evidence that the early development of cognitive deficits is significantly associated with PIGD or non-TD motor phenotypes [15,16], the motor phenotype of PD, particularly at a very early stage, might not yet be fully differentiated into two defined phenotypes (i.e., indeterminate type not specified as TD or PIGD) [10,17]. Therefore, whether the motor phenotype is useful in predicting the development of dementia in PD patients with very early stage disease and/or with PD medication should be further evaluated [17]. Among the clinical symptoms that progress heterogeneously, the predictability of cognitive decline in PD is currently evaluated by neuropsychological, neuropathologic, neuroimaging and biologic biomarkers. The neuropsychological assessment of PD patients will be a tool to help predict the development of dementia in PD patients. In fact, patients who meet the criteria for mild cognitive impairment are more likely to develop dementia [18,19]. The most common cognitive symptoms are attention deficits, executive functioning and visuospatial processing, and these variable profiles of cognitive dysfunction may be caused by the heterogeneous nature of the underlying neuropathology [19-21]. Several studies suggested that a specific domain of memory and cognitive function or the malfunctioning of a specific region were associated with a rapid progression of cognitive deficit, although it remains to be elucidated whether a specific profile reflects the heterogeneous progression of PD dementia [22]. For example, frontal cortex dysfunction assessed by executive difficulties is an early phenomenon, while visuospatial and semantic memory dysfunction, which reflect temporal and parietal involvement, are risk factors for Parkinson’s disease dementia (PDD).
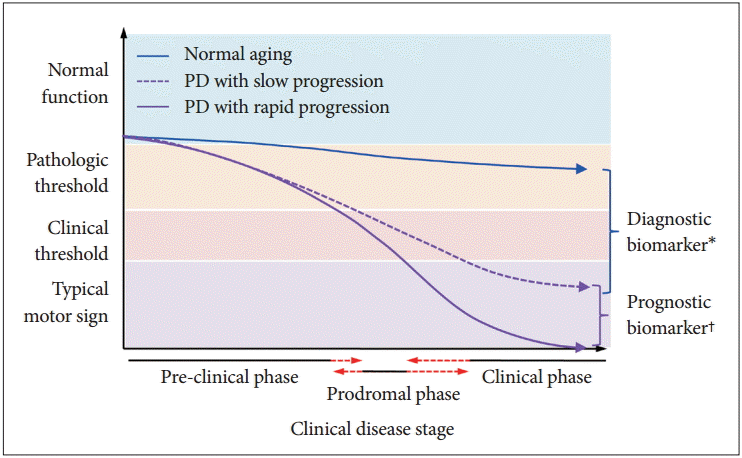
A hypothetical model of the heterogeneous progression of cognitive dysfunction in Parkinson’s disease (PD). *molecules involved in the pathogenesis of PD might be candidate diagnostic biomarkers that can be easily measured in biologic fluids. Several molecules, including α-synuclein, DJ-1, Fms-related tyrosine kinase 3 ligand, and proteome, were proposed as diagnostic biomarkers; however, their clinical performance in the diagnosis of PD was limited, †genetic, demographic, environmental, and medical conditions, such as concurrent diseases, might be factors that regulate the heterogeneous progression of PD. Genetic factors or protein molecules that regulate the progression of cognitive dysfunction in PD are candidate biomarkers of progression. However, longitudinal observation studies with large numbers of subjects in early stages of PD are required to discover valid progression biomarkers.
Neuropathologic substrates associated with cognitive heterogeneity
Although it is not sufficient, the measurement of CSF alpha-synuclein (α-syn), DJ-1, tau, phosphorylated tau at Thr181 (p-tau) or Fms-related tyrosine kinase 3 ligand can differentiate PD patients from healthy controls (HC). Indeed, the level of CSF α-syn and tau proteins in PD patients was significantly lower than those in age-matched HC, with a marked overlap between groups [17,23,24]. The mechanisms associated with lower levels of CSF α-syn in PD remain to be elucidated. The accumulation of α-syn in the brain–similar to amyloid β (Aβ) in Alzheimer’s disease (AD)–may be a mechanism for the reduction of CSF α-syn. Although it is necessary to characterize the immunoassay platform, the measurement of α-syn oligomers in CSF improves diagnostic utility [25]. Interestingly, the level of tau proteins [total tau (t-tau) and p-tau] in the CSF of PD patients was also lower than controls, although not all studies replicated this finding [26,27]. Taken together, the level of α-syn and other proteins in CSF may be useful in diagnosing PD, although it is too early to make definitive conclusions.
Braak et al. [28] revealed that the progression of α-syn pathology throughout the brain is not random but follows a stereotypical caudal-to-rostral ascending progression [29]; however, not all neuropathological studies agreed with that topology [30]. However, the detailed topographic patterns of the spread of α-syn among PD patients are not homogeneous. The specific topographical pattern of Lewy pathology (i.e., global involvement of cortical and limbic areas with Lewy bodies and Lewy neurites) is significantly associated with cognitive deficits in PD, which suggests the underlying dementia-prone pattern in the spread of Lewy pathology. Although it is not clear which factors regulate the topographic spread of Lewy pathology, human post-mortem studies suggested the cortical and limbic involvement of α-syn pathology as a neuropathologic substrate were strongly correlated with PDD [31,32]. Furthermore, recent advances in the understanding of the neuropathologic substrates of PDD suggested that the interaction between α-syn and AD pathology (i.e., Aβ and tau pathology) may regulate disease severity and progression [32-34]. Using CSF biomarkers, Siderowf et al. [35] provided evidence that the Alzheimer’s signature in CSF (lower level of CSF Aβ1-42) was significantly associated with more rapid cognitive decline in PD, and this finding was replicated by other studies [36-38]. The association of the AD-like CSF signature with the rapid progression of cognitive dysfunction supported the hypothetical model that cross-seeding α-syn and Aβ or tau accelerate neurodegenerative pathology and disease progression. In support of this assertion, transgenic mice overexpressing human α-syn (wild type or Ala53Thr mutant) with mutant forms of tau or amyloid precursor protein show greater neurodegeneration and functional deficits than mice overexpressing AD-associated proteins alone [39,40]. Furthermore, in vitro observation of enhanced fibril formation via cross-seed [41-43] and human studies in PD patients with the Ala53Thr SNCA mutation supported the contribution of AD pathology to the disease progression of PD [44]. The rapid progression of cognitive deficits in PD patients could not be exclusively determined by pathological substrates of AD; however, these previous studies in vitro, in animal models and in clinical cohorts suggested the increasingly important role of AD pathology in the development of PDD. For the prediction of cognitive progression in PD, genetic factors associated with neuropathological substrates of AD and/or PD may be an important contributor to cognitive deficits, along with α-syn pathology. For example, genetic factors (e.g., mutation in β-glucocerebrosidase) associated with the hereditary form of PD are a genetic predictor of PDD [45]. The apolipoprotein E (ApoE) E4 allele, which has been widely accepted as a risk factor for AD, may confer an increased risk of rapid cognitive decline in PD [46,47]. In contrast, the association of the ApoE ε4 allele with PDD was lost after adjustment for CSF Aβ1-42 level [35]. In a large-scale study with autopsied subjects, however, the ApoE ε4 allele frequency was significantly higher not only in the AD group but also in the dementia with Lewy bodies and PDD groups compared to the control group [48]. Therefore, the ApoE ε4 allele in PD may be a genetic risk factor for the cognitive deficits associated with pathways that are shared with and diverge from Aβ-related pathogenesis.
PARKINSON’S PROGRESSION MARKERS INITIATIVE
Overview
The development of reliable and validated biomarkers for heterogeneous PD progression is a critical unmet need. Validated PD progression markers are essential to accelerate research into PD pathogenesis, and the development of disease-modifying therapeutics (DMT) and would dramatically improve patient care. There are several important prerequisites for the development of valid biochemical progression biomarkers: the establishment of standardized protocols for the acquisition, transfer and analysis of biospecimens; the optimization and verification of bioassays; a sufficient longitudinal follow-up period to track heterogeneous progression; and the recruitment of drug-naïve patients at baseline. PPMI is a five-year observational, international, longitudinal study that aimed to identify biomarkers of PD progression that involve the collaborative effort of PD researchers with expertise in biomarker development, the clinical study of PD, bioinformatics, statistics and data management [11]. Analogous to the Alzheimer’s Disease Neuroimaging Initiative (ADNI), the PPMI is a public-private partnership, sponsored by the Michael J Fox Foundation with industry partnership. The overall objective of the PPMI study was to identify the clinical, imaging, and biologic markers of PD progression for use in clinical trials of DMT. Approximately 400 drug-naïve PD patients at the early stage, and 200 age-matched HC were planned to be enrolled from 24 clinical sites in the United States, Europe and Australia (Figure 2). The number of subjects was calculated with the power to detect a difference in prevalence of 13% (for a dichotomous endpoint) and a standardized mean difference of 0.24 (for a continuous end-point). All PD patients were at the early stage (diagnosis within 2 years and H&Y stage < 2) and untreated with PD medication, as described in detail elsewhere [11] and on the PPMI website (http://www.ppmi-info.org/study-design/). The longitudinal collection of biospecimens, including blood, CSF and urine, is an essential component to discovering biological markers that are able to track disease progression. In particular, the collection, processing, aliquoting and storage of CSF were remarkably standardized in the ADNI study. This review discusses the baseline CSF biomarkers (Aβ1-42, t-tau, p-tau, and α-syn) data of the PPMI cohort.
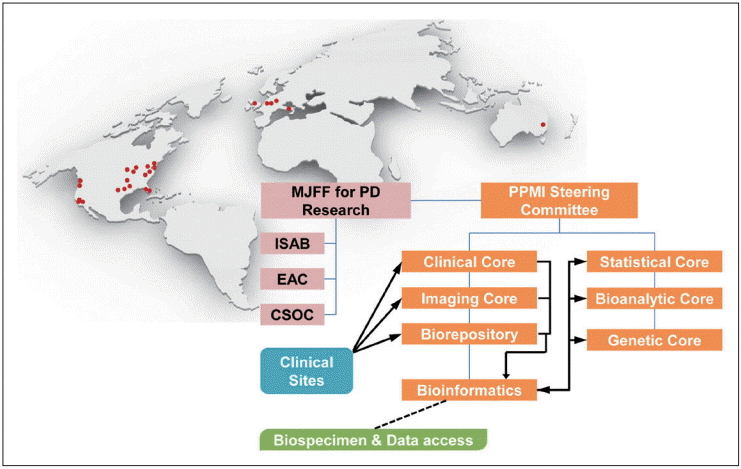
The organization and governance of the PPMI study and distribution of clinical sites in the United States, Europe and Australia. Data or samples obtained from each PPMI clinical site were transferred to clinical, imaging and biorepository cores, and all data were integrated in the PPMI study database. The PPMI data are publically available through the PPMI website (www.ppmi-info.org). Biological samples, including the longitudinal collection of CSF, blood and urine, were stored at the biorepository and are available to scientists via an application to the review committee through the website [11]. MJFF: Michael J. Fox Foundation, ISAB: Industry Scientific Advisory Board, EAC: External Advisory Committee, CSOC: Clinical Study Oversight Committee, PD: Parkinson’s disease, PPMI: Parkinson Progression Marker Initiative, CSF: cerebrospinal fluid.
Association of CSF biomarkers with clinical features
The partial baseline CSF results (n = 102; PD = 63, HC = 39) were published in 2013 [17]. The initial data showed several interesting findings; the lower levels of CSF Aβ1-42 and p-tau were significantly associated with the PIGD phenotype in multiple logistic regression analysis with adjustment for confounders; the level of α-syn was significantly correlated with the level of t-tau and p-tau, and the levels of α-syn and t-tau were associated with motor severity. A recent analysis of the full baseline dataset showed consistent results (n = 660; PD = 412, HC = 189, subjects without evidence of dopamine deficit = 59), but some results could not replicate the pilot findings [10]. For example, the level of CSF Aβ1-42 or p-tau was not associated with motor phenotype, but the CSF α-syn level in PD patients with the non-TD phenotype was significantly lower than PD patients with the TD phenotype. In addition, there were no CSF biomarkers that were significantly associated with motor severity when multivariate regression analysis with the adjustment of confounding factors was applied, although a low level of p-tau was marginally associated with disease severity. However, the strong correlation between the level of α-syn and t-tau or p-tau in both PD and HC was replicable. Consistent with the pilot study, the levels of CSF α-syn, t-tau, and p-tau, but not Aβ1-42, were significantly lower in PD compared to HC, while the diagnostic utility of each biomarker was limited due to a large overlap. The lower level of CSF α-syn in PD relative to HC implicates the accumulation of α-syn in the brain of PD patients, analogous to the finding of lower levels of CSF Aβ1-42 in AD patients compared to HC. The mechanism of a reduction in tau proteins in PD compared with HC is unclear; however, a possible interpretation is that the interaction between tau proteins and α-syn may limit the release of tau proteins into CSF. In connection with this, previous studies using in vitro [49], animal models [49-51] or postmortem brains of PD [52,53] reported that the α-syn pathology in the brain is accompanied by increased levels of hyperphosphorylated tau proteins and tau-positive tangles, and α-syn positive Lewy bodies may co-localize in the same neuron [54]. The genome-wide association study also supported this hypothesis that MAPT and SNCA, which encode tau and α-syn, respectively, showed a genetic association with PD [55]. Therefore, the extent of the direct or indirect interaction between tau phosphorylation and α-syn accumulation or the pattern of topological distribution of these pathogenic proteins may contribute to the heterogeneous progression of PD. However, the mechanisms that regulate the interaction of α-syn with tau are unclear. Although future long-term longitudinal observations in the PPMI cohort will be required to test the predictive performance of the CSF biomarkers, the baseline data in this large cohort suggest that CSF biomarkers in early PD patients already reflect disease heterogeneity and may have predictive value for disease progression. Our findings of the association of CSF biomarkers with cognitive function in the PPMI cohort was not consistent with other studies [26,35,37,56-60]. For example, the association of a higher CSF α-syn level with worsening cognitive decline was observed in the Deprenyl and tocopherol antioxidative therapy of parkinsonism study [56,57]. In contrast, the lower CSF α-syn level was associated with more severe neuropsychological function, including semantic fluency, visuospatial cognition and executive functioning in the PPMI cohort, which indicated that α-syn pathology contributes to early cognitive impairment in PD. In addition, multivariate regression analysis of the PPMI baseline data did not fully reproduce the previous findings that the lower level of CSF Aβ1-42 was associated with cognitive impairment in PD [26,35,59,60]. The lower level of Aβ1-42 was significantly associated with processing speed/attention assessed by the Symbol Digit Modality Test (SDMT) but not with other cognitive functions, including the global cognitive function test and Montreal Cognitive Assessment, in the PPMI cohort. Instead, when the clinical variables of the group with the highest quintile levels of CSF biomarkers were compared with those of the group with the lowest quintile levels, the CSF Aβ1-42 level showed significant associations with semantic fluency and SDMT score, and the t-tau/Aβ1-42 ratio showed significant associations with memory (total recall and delayed recall measured by Hopkins Verbal Learning Test-Revised score), semantic fluency, SDMT and Wechsler Memory ScaleIII Letter-Number Sequencing score. It should be noted that the PPMI cohort included patients with very early stage and drug-naïve disease at baseline; therefore, whether CSF biomarkers in early PD are associated with the risk of future cognitive decline and PDD should be determined in longitudinal analyses. A recent study that observed a group of PPMI PD patients (n = 341) for 2 years found a significant association of lower baseline CSF Aβ1-42 level with higher odds of cognitive impairment [61], even though the baseline CSF biomarker data showed a slight association of CSF Aβ1-42 with cognitive dysfunction in multivariate analysis. The discrepancy among studies in the association of CSF biomarkers with clinical variables may be due to several demographic, biological and analytical factors, including but not limited to the different ages among cohorts, the contamination of blood in CSF, the mixed pathology or disease stage of studied patients, and different immunoassay platforms [62]. Therefore, we should carefully interpret the results for the association of CSF biomarkers with clinical variables.
PERSPECTIVE
Not all PD patients develop dementia; however, dementia is a frequent non-motor complication in PD with heterogeneous features. Genetic, demographic and environmental factors may be related to the heterogeneous progression of cognitive decline in PD patients. Thus, understanding the heterogeneity would provide insight into the pathogenic mechanism of PDD development, which is important for developing therapeutics as well as patient care. There are few studies on the association of heterogeneous cognitive decline with specific molecular signatures or pathogenesis in PD patients with a large number of subjects and in early stages of the disease [17,23,26,58]. The evidence of molecular interactions between α-syn and Aβ1-42 and/or tau in the development of PDD in post-mortem samples implicate the necessity of the longitudinal observation of CSF and imaging biomarkers in PD. In addition, the development of biomarkers for the early diagnosis of PD is an unmet need. Because PD is not a disease with homogeneous features, the development of biomarkers of the heterogeneity in motor and non-motor dysfunction will provide the molecular basis for individualized therapeutics. Finally, the combination of biochemical, imaging and genetic biomarkers rather than individual biomarkers may better accomplish this aim. To this end, the development of novel, promising and valid biochemical biomarkers and α-syn imaging technologies is necessary.
Notes
Conflicts of Interest
The author has no financial conflicts of interest.
Acknowledgements
This study was supported by the Mid-Career Researcher Program (2013R1A2A2A01008223) through the National Research Foundation of Korea (NRF).