ABSTRACT
- Episodic ataxia (EA) is a clinically heterogeneous group of disorders that are characterized by recurrent spells of truncal ataxia and incoordination lasting minutes to hours. Most have an autosomal dominant inheritance pattern. To date, 8 subtypes have been defined according to clinical and genetic characteristics, and five genes are known to be linked to EAs. Both EA1 and EA2, which are caused by mutations in KCNA1 and CACNA1A, account for the majority of EA, but many patients with no identified mutations still exhibit EA-like clinical features. Furthermore, genetically confirmed EAs have mostly been identified in Caucasian families. In this article, we review the current knowledge on the clinical and genetic characteristics of EAs. Additionally, we summarize the phenotypic features of the genetically confirmed EA2 families in Korea.
-
Keywords: Episodic ataxia; KCNA1; CACNA1A
INTRODUCTION
- Episodic ataxia (EA) is a clinically heterogeneous group of disorders that are characterized by recurrent spells of truncal ataxia and incoordination [1,2]. The incidence is likely to be less than 1/100,000, but it may be underestimated due to demanding genetic tests and unidentified causative genes. Most have an autosomal dominant inheritance pattern, although some sporadic cases have been reported. There are several subtypes of EA, and these subtypes are defined according to clinical features and genetic characterizations. However, clinical and genetic heterogeneity is not uncommon (Table 1). Only EA1 and EA2 have been reported in multiple families of different ethnicity. They are caused by mutations involving the potassium and calcium channel genes, KCNA1 and CACNA1A. Mutations in CACNB4 and SLC1A3 are known to cause EA5 and EA6, respectively, but have been reported in only one or two families [3-5]. Recently, by whole-exome sequencing, UBR4 was found to be associated with EA [6].
- In Korea, genetically confirmed EA has rarely been reported, probably due to clinical heterogeneity and the limited availability of commercial genetic tests [7-10]. However, recent advances in molecular genetics have led to the increased identification of candidate variants and new mutations associated with EAs.
- This review introduces the updates on the clinical and genetic features of EAs and the phenotypes of Korean EA patients that have been reported in the literature.
CLINICAL FEATURES
- The key clinical feature of EAs is the discrete attacks of incoordination, with a clear onset and resolution of symptoms [1,2]. However, some patients can also have progressive ataxia, which makes it difficult to distinguish EA with progressive features from progressive ataxia with intermittent exacerbation. Furthermore, the clinical features of EA subtypes overlap each other. Among the various subtypes, only EA1 and EA2 have the characteristic interictal findings and well-defined clinical features.
- EA1
- EA1 is characterized by brief episodes of ataxia with constant interictal myokymia [1,2,11,12]. The onset is typically during early childhood. The core features are imbalance, incoordination and slurred speech and are triggered by physical and emotional stress, startle response, or abrupt movements. The typical duration of attacks lasts seconds to minutes, but prolonged attacks lasting hours or days have also been described [13]. Furthermore, a recent large prospective study revealed that at least one-fifth of EA1 patients developed permanent cerebellar symptoms and signs [11]. Myokymia may be clinically evident or only detectable by electromyography. Neuromyotonia is also apparent in most patients, to a varying degree, and is characterized by muscle stiffness, twitching, flickering, and muscle hypertrophy. The presence of myokymia or neuromyotonia suggests the involvement of the peripheral nervous system. Additional symptoms include distal weakness, tremors, choreoathetosis, spinning sensation, and cognitive dysfunctions.
- EA2
- EA2 is the most common subtype of EA. The episodes are characterized by recurrent ataxia, slurred speech for several hours, and interictal nystagmus. The onset is typically early in life, but patients with an onset during the sixth decade have also been reported [8,14,15]. Vertigo and fluctuating general weakness are common. Additional features include diplopia, tinnitus, seizure, dystonia, and cognitive impairment. Some patients initially present with a paroxysmal tonic upward gaze lasting a few minutes to one hour during infancy, and the patients then gradually develop cerebellar symptoms as the tonic upward gaze disappears [16,17]. Similarly, in EA1, the attacks are commonly triggered by exercise, or physical or emotional stress. A recent study described that severe postural instability with pure downbeat nystagmus was provoked by intense exercise in a patient with EA2 [8]. Between the attacks, patients may be free of symptoms but can present mildly progressive baseline ataxia. Most patients also show various interictal nystagmus, such as gaze-evoked nystagmus (GEN), rebound nystagmus, or primary position downbeat nystagmus. Acetazolamide can prevent the attacks by reducing arterial pH, but it may cause kidney stones. Alternatively, aminopyridine, a nonselective blocker of voltage-gated potassium channels, reduces the frequency of attacks and improves the quality of life in patients with EA2 [18].
- Other EA subtypes
- Other EA subtypes have only been reported in one or two families.
- EA3 was described in a single large Canadian family [19]. They were characterized by recurrent episodes of truncal ataxia, vertigo, and tinnitus, usually lasting less than 30 minutes. Other features included interictal myokymia, headache, visual blurring, diplopia, and weakness. The presence of vertigo and tinnitus and the absence of interictal nystagmus and shorter episodes distinguish EA3 from EA1 and EA2.
- EA4, which is also called periodic vestibulocerebellar ataxia, was described in two North Carolina kindreds, suggesting a single common founder [20,21]. They were characterized by ataxia, vertigo, GEN, and defective smooth pursuit. The age of onset ranged from the third to sixth decades. The attacks typically last hours and do not response to acetazolamide.
- EA5, which is caused by a mutation in CACNB4, was reported in a French Canadian family [3]. The clinical features were similar to those of EA2, including the duration of attacks, interictal nystagmus, and response to acetazolamide. The only obvious difference is that its onset is later than that of EA2.
- EA6 is caused by a mutation in SLC1A3 and was identified in two unrelated individuals [4,5]. Their clinical phenotypes were different from each other. One sporadic child presented with a severe form of EA with seizure, migraine, and alternating hemiplegia, which was triggered by febrile illness [4]. The other Dutch family showed typical EA2-like symptoms, such as episodes with a duration of several hours, interictal nystagmus, and a positive response to acetazolamide [5].
- EA7 was reported in a 4-generation family, in which 7 members had EA [22]. The clinical features were similar to those of EA2, except without interictal findings.
- Recently, the Online Mendelian Inheritance of Man database registered a large 3-generation Irish family with EA as EA8 [6]. The attacks were characterized by unsteadiness, general weakness, and slurred speech. Additional features included twitching around the eyes, nystagmus, myokymia, and persistent intention tremor. An interesting finding was the response to clonazepam, instead of acetazolamide, which was consistent within the affected individuals.
GENETIC CHARACTERISTICS
- There are at least 8 loci for EA, 5 of which are known genes (Table 1). All of the identified genes, except UBR4, encode ion channel proteins located on the neuronal or glial membrane and play important roles in excitatory neurotransmission.
- EA1: KCNA1
-
KCNA1, which is located on chromosome 12p13, is the gene responsible for EA1 [1,2,11,12]. It encodes the voltage-gated potassium channel, Kv1.1, which is expressed highly in basket cells and interneurons forming the GABAergic synapses on Purkinje cells. Kv1.1 plays an important role in the repolarization phase of presynaptic action potentials that affect the inhibitory inputs to the Purkinje cells. Thus, KCNA1 mutations result in the hyperexcitability of the presynaptic basket cells and the excessive release of the neurotransmitter, GABA, which can inhibit the generation of action potentials in the Purkinje cells. As a consequence, the inhibitory output of the cerebellum may be markedly reduced, thus producing the cerebellar symptoms observed in EA1 patients.
- To date, 30 mutations have been identified in EA1 individuals, and the majority are missense point mutations [1,11,12]. Four different mutations have been described on the highly conserved threonine residue at codon 226 (T226M/A/R/K), which is located in the second transmembrane segment, but the phenotypic characteristics are diverse. This can be explained by the extent of functional impairment related to the potassium channel, depending on the type of KCNA1 mutation. In other words, all mutations result in loss-of-function effects on the outward K+ flux by altering channel expression and gating, but the effect of each mutation is highly heterogeneous. Furthermore, due to wide interfamilial and intrafamilial phenotypic variability, genotype-phenotype correlations are difficult to establish [11].
- EA2: CACNA1A
- EA2 is caused by mutations involving CACNA1A on chromosome 19p13, which encodes Cav2.1, the α1 subunit of the P/Q-type voltage-gated calcium channel [1,2]. This channel is widely expressed in the Purkinje and granule cells of the cerebellum; it mediates calcium entry into the cells and regulates the precision of pacemaking. Thus, CACNA1A mutations lead to a decrease in Ca2+ entry through Cav2.1 and the irregular firing of the Purkinje cells, which contributes to EA2 symptoms.
- Over 60 different heterozygous point mutations have been described in EA2 patients. Most mutations disrupt the open reading frame and subsequently lead to a premature stop due to nonsense mutation or defects in splice sites. However, some missense mutations have also been reported. In some EA2 patients, the direct sequencing of CACNA1A did not identify any point mutation, but the use of multiplex ligation-dependent probe amplification demonstrated large genomic deletions or duplications [23,24]. Although missense mutations typically involve the pore loop region of the protein, CACNA1A mutations occur throughout the entire gene, without any consistent hot spots.
-
CACNA1A is also the gene responsible for familial hemiplegic migraine type 1 (FHM1) and spinocerebellar ataxia type 6 (SCA6). However, in FHM1 and SCA6, increased Ca2+ influx through the mutant channel causes aberrant neurotransmitter release and excitotoxicity [1]. Nevertheless, there is clinical overlap among EA2, FHM1, and SCA6. Many patients with FHM1 show cerebellar symptoms and signs, whereas over half of the EA2 patients have migraines that meet the International Headache Society criteria. Some patients with SCA6 also present with fluctuating ataxia similar to EA2.
- Other EAs
- In addition to KCNA1 and CACNA1A, there are 3 other EA genes.
- EA5 results from mutations involving CACNB4, which encodes the calcium channel β4 subunit and is located on chromosome 2q22-23 [3]. This subunit interacts directly with the C-terminus of α1 subunit (Cav2.1) and contributes to the modulation of calcium current amplitude, voltage dependence, and kinetics of activation and inactivation. A missense mutation C104F has been identified in an EA5 family, but the functional alterations of channel kinetics were subtle. CACNB4 mutations have also been reported in a family with generalized epilepsy (C104F) and juvenile myoclonic epilepsy (R482X).
- EA6 is caused by mutations in SLC1A3 on chromosome 5p, which encodes excitatory amino acid transporter 1 (EAAT1), a glial glutamate transporter [4,5]. EAAT1 is present in the brainstem and cerebellum and is responsible for glutamate uptake in the synapses. Thus, SLC1A3 mutation leads to the excessive extracellular accumulation of glutamate and neurotoxic insults. Only 2 mutations, P290R and C186S, have been reported in the literature, and the mutant EAAT1 has been shown to result in decreased glutamate uptake in cell culture assays.
- Recently, the missense variant, R5091H, in UBR4, which is located on chromosome 1p36.13, was found in a large Irish family with autosomal dominant EA8 by whole-exome sequencing [6]. However, its contribution to EA8 has not been confirmed by functional studies. UBR4 is known to interact with calmodulin, a Ca2+ regulating protein in the cytoplasm, and may thus be involved in the regulation of neuronal excitability.
- Although recent advances in next-generation sequencing techniques have contributed to the easy identification of pathogenic mutations, many patients with typical clinical features of EA do not have any mutations in the known EA genes. This suggests a genetic heterogeneity of EA and the presence of additional causative EA genes. EA3 and EA7 are linked to chromosome 1q42 and 19q13, respectively, but no candidate gene has been confirmed [19,22]. In EA4, linkage analysis ruled out EA1 and EA2 loci, but to date, no genome-wide scan has been reported [21].
- Many genes coding the ion channels, transporters, or synaptic proteins play crucial roles in regulating neural excitability in the central nervous system. Mutations in these genes can cause various paroxysmal neurological symptoms, and several overlapping syndromes have been described, of which EA has been reported as one of various phenotypes (Table 2). For example, mutations in SCN2A, encoding the voltage-gated Na+ channel, Nav 1.2, cause benign familial neonatal-infantile seizures but can also contribute to later onset EA [25,26]. Other genes associated with EA include ATP1A3 [27], NALCN [28], DARS2 [29], SLC2A1 [30], FGF14 [31], and PRRT2 [32], and they have been reported to cause epilepsy and paroxysmal dyskinesia. Thus, extensive searches for these genes with whole-exome sequencing may help define novel candidate genes and identify new mutations associated with EA.
EAS IN KOREA
- To date, four Korean EA2 families have been reported in the literature (Table 3, Figure 1) [7-10]. Two splice site mutations, one nonsense mutation and one missense mutation involving CACNA1A were identified, and one of them was previously reported as a pathogenic mutation in a Caucasian EA2 patient [33]. The clinical features of the Korean patients were similar to those of Caucasian EA2 patients, including recurrent episodes of ataxia or vertigo lasting hours, interictal nystagmus and a good response to acetazolamide. However, each family had an intriguing feature. In the first family with a deletion mutation (c.2042_2043delAG), the symptoms were provoked by a change in body temperature, such as heat or high fever [7]. This suggests that calcium channel function may have a strong dependence on body temperature. The second family with a nonsense mutation (c.3832C>T) presented with recurrent ataxia and vertigo that were frequently triggered by exercise, and one member developed pure downbeat nystagmus and severe postural imbalance after exercise [8]. Ictal downbeat nystagmus may be ascribed to changes in the cerebellar pH homeostasis precipitated by hyperventilation during the exercise. The third family with a splice site mutation (c.4392-1G>C) showed a decrease in the age of onset, possibly due to genetic anticipation, in three succeeding generation [9]. Anticipation mostly occurs in neurodegenerative diseases that are characterized by an expansion of unstable trinucleotide repeats, but it has not been reported in EA2. In the last family with another splice site mutation (c.4953+1G>A), one patient showed transient upbeat nystagmus on resuming primary eye position after lateral gazes that had induced GEN and downbeat nystagmus [10]. This phenomenon can be explained by a shifting null in the vertical planes as a result of an adaptation to the downbeat nystagmus that developed during lateral gaze.
- Although only four Korean EA2 families have been reported in the literature and other subtypes have not yet been identified, this does not mean that the frequency of EA is lower in the Korean population compared to Caucasian populations. Although there are many clinically diagnosed EA patients without genetic confirmations in multiple centers, the limited availability of genetic tests make it difficult to establish a genetic confirmation. As whole-exome sequencing becomes more affordable, the identification of new mutations associated with EAs will increase in the future.
Notes
-
Conflicts of Interest
The authors have no financial conflicts of interest.
Acknowledgments
- This research was supported by Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education (NRF-2015R1D1A1A01060559).
Figure 1.Mutations of the α1 subunit of the P/Q-type voltage-gated calcium channel in Korean patients with episodic ataxia type 2. The protein contains four homologous domains (I–IV), each with six transmembrane segments (S1–S6). The numbers in the symbol correspond to the mutations listed in Table 3.
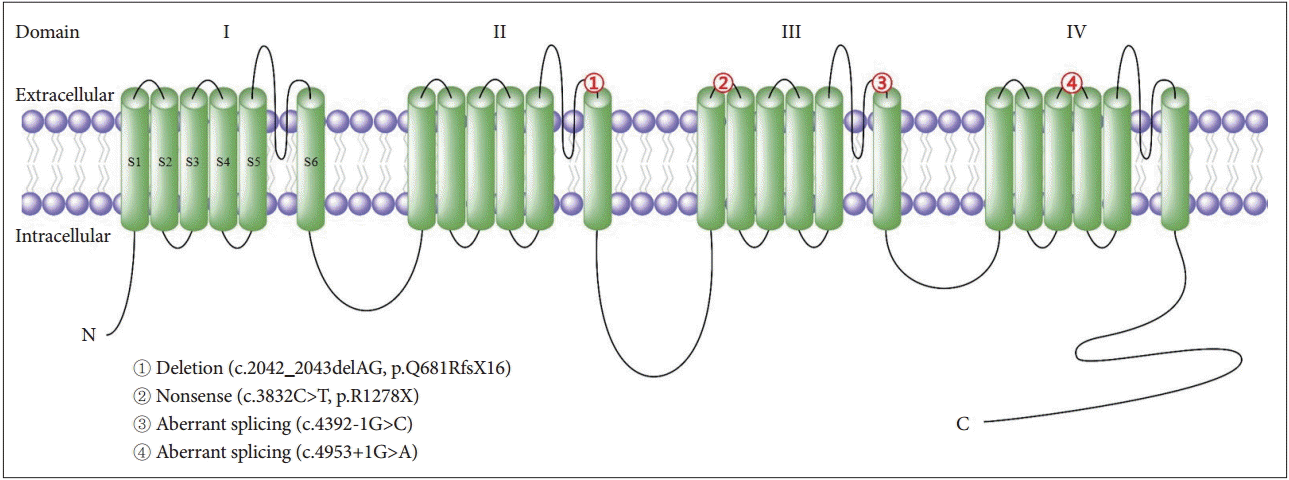
Table 1.Genetic and clinical summary of episodic ataxia (EA)
|
Type |
OMIM |
Inheritance |
Genes |
Protein |
Age of onset |
Attack duration |
Associated symptoms |
Interictal findings |
|
EA1 |
160120 |
AD |
KCNA1 (12p13) |
Potassium channel (Kv1.1) |
2-15 |
Seconds-min |
Vertigo, dysarthria, weakness, tremor, seizure |
Myokymia |
|
EA2 |
108500 |
AD |
CACNA1A (19p13) |
P/Q type calcium channel |
2-20 |
Hours |
Vertigo, dysarthria, diplopia, weakness, tonic upward gaze, headache, seizure, dystonia, cognitive impairment |
Nystagmus, ataxia |
|
α1 subunit (Cav2.1) |
|
EA3 |
606554 |
AD |
Unknown |
Unknown |
1-42 |
1 min to 6 h |
Vertigo, diplopia, weakness, tinnitus, headache, visual blurring |
Myokymia |
|
EA4 |
606552 |
AD |
Unknown |
Unknown |
23-60 |
Brief |
Vertigo, diplopia |
Nystagmus, abnormal smooth pursuit |
|
EA5 |
613855 |
AD |
CACNB4 (2q22-23) |
P/Q type calcium channel |
> 20 |
Hours |
Vertigo, dysarthria |
Nystagmus, ataxia |
|
β4 subunit |
|
EA6 |
612656 |
AD or sporadic |
SLC1A3 (5p13.2) |
Excitatory amino acid transporter 1 |
5-14 |
Hours-days |
Vertigo, weakness, seizure |
Nystagmus, ataxia |
|
EA7 |
611907 |
Multiple |
Unknown |
Unknown |
< 20 |
Hours-days |
Vertigo, dysarthria, weakness |
No |
|
EA8 |
616055 |
AD |
UBR4 (1p36.13) |
Ubiquitin-protein ligase |
Early infancy |
Min to 24 h |
Vertigo, weakness |
Nystagmus, ataxia myokymia |
Table 2.Other genes associated with episodic ataxia (EA)
|
Genes |
Locus |
Protein |
Clinical phenotype |
Reference |
|
SCN2A
|
2q24.3 |
Na+ channel (Nav 1.2) |
Neonatal epilepsy, late-onset episodic ataxia, myoclonus, and pain |
[25, 26] |
|
Benign familial neonatal-infantile seizure |
|
|
ATP1A3
|
19q13.2 |
Na+/K+ ATPase |
Episodic cerebellar ataxia, areflexia, optic atrophy, sensorineural hearing loss (CAOS) |
[27] |
|
NALCN
|
13q32.3 |
Na+ leak channel |
Intellectual disability, acetazolamide-responsive episodic ataxia |
[28] |
|
Congential contractures of the limbs and face, hypotonia, and developmental delay |
|
|
DARS2
|
1q25.1 |
Aspartyl-t RNA synthetase |
Acetazolamide-responsvie exercise-induced episodic ataxia |
[29] |
|
Leukoencephalopathy with brainstem and spinal cord involvement and brain lactate elevation |
|
|
SLC2A1
|
1p34.2 |
Glucose transporter 1 |
Intermittent ataxia, similar to EA2 |
[30] |
|
Dystonia, GLUT1 deficiency syndrome |
|
|
FGF14
|
13q33.1 |
Fibroblast growth factor 14 |
Episodic ataxia |
[31] |
|
Spinocerebellar ataxia 27 |
|
|
PRRT2
|
16p11.2 |
Proline-rich transmembrane protein 2 |
Convulsions, familial infantile with paroxysmal choreoathetosis |
[32] |
|
Episodic kinesigenic dyskinesis 1 |
|
|
Seizure, benign familial infantile 2 |
|
Table 3.Literature reports of Korean families with episodic ataxia type 2
|
Family |
CACNA1A mutation (NM_001127221.1) |
Domain |
Affected members |
Age of onset |
Ictal symptoms |
Interictal nystagmus |
Additional features |
Response to acetazolamide |
Reference |
|
1 |
Exon 16, deletion, c.2042_2043delAG, p.Q681RfsX16 |
II S5-S6 |
3 |
10-15 |
Ataxia, dysarthria |
GEN |
Provoked by a heat |
1/1 |
[7] |
|
2 |
Exon 23, nonsense c.3832C>T, p.R1278X |
III S1-S2 |
4 |
12-25 |
Ataxia, vertigo, dysarthria, headache, paresthesia |
(-) |
Exercise-induced downbeat nystagmus |
1/1 |
[8] |
|
3 |
Intron 27, aberrant splicing, c.4392-1G>C |
III S5-S6 |
6 |
5-56 |
Ataxia, vertigo, dysarthria |
GEN, DB |
Possible anticipation |
2/2 |
[9] |
|
4 |
Intron 31, aberrant splicing, c.4953+1G>A |
IV S3-S4 |
3 |
10-12 |
Ataxia, vertigo, dysarthria, headache, |
GEN, DB |
Rebound upbeat nystagmus |
1/1 |
[10] |
REFERENCES
- 1. Jen JC, Graves TD, Hess EJ, Hanna MG, Griggs RC, Baloh RW; CINCH investigators. Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain 2007;130(Pt 10):2484–2493.ArticlePubMedPDF
- 2. Tomlinson SE, Hanna MG, Kullmann DM, Tan SV, Burke D. Clinical neurophysiology of the episodic ataxias: insights into ion channel dysfunction in vivo. Clin Neurophysiol 2009;120:1768–1776.ArticlePubMed
- 3. Escayg A, De Waard M, Lee DD, Bichet D, Wolf P, Mayer T, et al. Coding and noncoding variation of the human calcium-channel beta4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am J Hum Genet 2000;66:1531–1539.ArticlePubMedPMC
- 4. Jen JC, Wan J, Palos TP, Howard BD, Baloh RW. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology 2005;65:529–534.ArticlePubMed
- 5. de Vries B, Mamsa H, Stam AH, Wan J, Bakker SL, Vanmolkot KR, et al. Episodic ataxia associated with EAAT1 mutation C186S affecting glutamate reuptake. Arch Neurol 2009;66:97–101.ArticlePubMed
- 6. Conroy J, McGettigan P, Murphy R, Webb D, Murphy SM, McCoy B, et al. A novel locus for episodic ataxia: UBR4 the likely candidate. Eur J Hum Genet 2014;22:505–510.ArticlePubMedPMC
- 7. Kim JM, Kim JS, Ki CS, Jeon BS. Episodic ataxia type 2 due to a deletion mutation in the CACNA1A gene in a Korean family. J Clin Neurol 2006;2:268–271.ArticlePubMedPMC
- 8. Choi JH, Seo JD, Choi YR, Kim MJ, Shin JH, Kim JS, et al. Exercise-induced downbeat nystagmus in a Korean family with a nonsense mutation in CACNA1A. Neurol Sci 2015;36:1393–1396.ArticlePubMed
- 9. Choi KD, Yook JW, Kim MJ, Kim HS, Park YE, Kim JS, et al. Possible anticipation associated with a novel splice site mutation in episodic ataxia type 2. Neurol Sci 2013;34:1629–1632.ArticlePubMed
- 10. Kim HJ, Kim JS, Choi JH, Shin JH, Choi KD, Zee DS. Rebound upbeat nystagmus after lateral gaze in episodic ataxia type 2. Cerebellum 2014;13:411–413.ArticlePubMed
- 11. Graves TD, Cha YH, Hahn AF, Barohn R, Salajegheh MK, Griggs RC, et al. Episodic ataxia type 1: clinical characterization, quality of life and genotype-phenotype correlation. Brain 2014;137(Pt 4):1009–1018.ArticlePubMedPMCPDF
- 12. D’Adamo MC, Hasan S, Guglielmi L, Servettini I, Cenciarini M, Catacuzzeno L, et al. New insights into the pathogenesis and therapeutics of episodic ataxia type 1. Front Cell Neurosci 2015;9:317.ArticlePubMedPMC
- 13. Lee HY, Xu Y, Huang Y, Ahn AH, Auburger GW, Pandolfo M, et al. The gene for paroxysmal non-kinesigenic dyskinesia encodes an enzyme in a stress response pathway. Hum Mol Genet 2004;13:3161–3170.ArticlePubMedPDF
- 14. Imbrici P, Eunson LH, Graves TD, Bhatia KP, Wadia NH, Kullmann DM, et al. Late-onset episodic ataxia type 2 due to an in-frame insertion in CACNA1A. Neurology 2005;65:944–946.ArticlePubMed
- 15. Cuenca-León E, Banchs I, Serra SA, Latorre P, Fernàndez-Castillo N, Corominas R, et al. Late-onset episodic ataxia type 2 associated with a novel loss-of-function mutation in the CACNA1A gene. J Neurol Sci 2009;280:10–14.ArticlePubMed
- 16. Roubertie A, Echenne B, Leydet J, Soete S, Krams B, Rivier F, et al. Benign paroxysmal tonic upgaze, benign paroxysmal torticollis, episodic ataxia and CACNA1A mutation in a family. J Neurol 2008;255:1600–1602.ArticlePubMed
- 17. Blumkin L, Leshinsky-Silver E, Michelson M, Zerem A, Kivity S, Lev D, et al. Paroxysmal tonic upward gaze as a presentation of de-novo mutations in CACNA1A. Eur J Paediatr Neurol 2015;19:292–297.ArticlePubMed
- 18. Strupp M, Kalla R, Dichgans M, Freilinger T, Glasauer S, Brandt T. Treatment of episodic ataxia type 2 with the potassium channel blocker 4-aminopyridine. Neurology 2004;62:1623–1625.ArticlePubMed
- 19. Steckley JL, Ebers GC, Cader MZ, McLachlan RS. An autosomal dominant disorder with episodic ataxia, vertigo, and tinnitus. Neurology 2001;57:1499–1502.ArticlePubMed
- 20. Farmer TW, Mustian VM. Vestibulocerebellar ataxia. A newly defined hereditary syndrome with periodic manifestations. Arch Neurol 1963;8:471–480.ArticlePubMed
- 21. Damji KF, Allingham RR, Pollock SC, Small K, Lewis KE, Stajich JM, et al. Periodic vestibulocerebellar ataxia, an autosomal dominant ataxia with defective smooth pursuit, is genetically distinct from other autosomal dominant ataxias. Arch Neurol 1996;53:338–344.ArticlePubMed
- 22. Kerber KA, Jen JC, Lee H, Nelson SF, Baloh RW. A new episodic ataxia syndrome with linkage to chromosome 19q13. Arch Neurol 2007;64:749–752.ArticlePubMed
- 23. Wan J, Mamsa H, Johnston JL, Spriggs EL, Singer HS, Zee DS, et al. Large genomic deletions in CACNA1A cause episodic ataxia type 2. Front Neurol 2011;2:51.ArticlePubMedPMC
- 24. Riant F, Mourtada R, Saugier-Veber P, Tournier-Lasserve E. Large CACNA1A deletion in a family with episodic ataxia type 2. Arch Neurol 2008;65:817–820.ArticlePubMed
- 25. Liao Y, Anttonen AK, Liukkonen E, Gaily E, Maljevic S, Schubert S, et al. SCN2A mutation associated with neonatal epilepsy, late-onset episodic ataxia, myoclonus, and pain. Neurology 2010;75:1454–1458.ArticlePubMed
- 26. Schwarz N, Hahn A, Bast T, Müller S, Löffler H, Maljevic S, et al. Mutations in the sodium channel gene SCN2A cause neonatal epilepsy with late-onset episodic ataxia. J Neurol 2016;263:334–343.ArticlePubMedPDF
- 27. Heimer G, Sadaka Y, Israelian L, Feiglin A, Ruggieri A, Marshall CR, et al. CAOS-episodic cerebellar ataxia, areflexia, optic atrophy, and sensorineural hearing loss: a third allelic disorder of the ATP1A3 gene. J Child Neurol 2015;30:1749–1756.ArticlePubMed
- 28. Aoyagi K, Rossignol E, Hamdan FF, Mulcahy B, Xie L, Nagamatsu S, et al. A gain-of-function mutation in NALCN in a child with intellectual disability, ataxia, and arthrogryposis. Hum Mutat 2015;36:753–757.ArticlePubMed
- 29. Synofzik M, Schicks J, Lindig T, Biskup S, Schmidt T, Hansel J, et al. Acetazolamide-responsive exercise-induced episodic ataxia associated with a novel homozygous DARS2 mutation. J Med Genet 2011;48:713–715.ArticlePubMed
- 30. Ohshiro-Sasaki A, Shimbo H, Takano K, Wada T, Osaka H. A three-year-old boy with glucose transporter type 1 deficiency syndrome presenting with episodic ataxia. Pediatr Neurol 2014;50:99–100.ArticlePubMed
- 31. Choquet K, La Piana R, Brais B. A novel frameshift mutation in FGF14 causes an autosomal dominant episodic ataxia. Neurogenetics 2015;16:233–236.ArticlePubMed
- 32. Gardiner AR, Bhatia KP, Stamelou M, Dale RC, Kurian MA, Schneider SA, et al. PRRT2 gene mutations: from paroxysmal dyskinesia to episodic ataxia and hemiplegic migraine. Neurology 2012;79:2115–2121.ArticlePubMedPMC
- 33. Yue Q, Jen JC, Thwe MM, Nelson SF, Baloh RW. De novo mutation in CACNA1A caused acetazolamide-responsive episodic ataxia. Am J Med Genet 1998;77:298–301.ArticlePubMed
Citations
Citations to this article as recorded by

- Integration of multi-omics technologies for molecular diagnosis in ataxia patients
Sebastien Audet, Valerie Triassi, Myriam Gelinas, Nab Legault-Cadieux, Vincent Ferraro, Antoine Duquette, Martine Tetreault
Frontiers in Genetics.2024;[Epub] CrossRef - Genetics and Pathogenesis of Dystonia
Mirja Thomsen, Lara M. Lange, Michael Zech, Katja Lohmann
Annual Review of Pathology: Mechanisms of Disease.2024; 19(1): 99. CrossRef - Episodic ataxia type 2 with a novel missense variant (Leu602Arg) in CACNA1A
Shiroh Miura, Emina Watanabe, Kensuke Senzaki, Shigeyoshi Hiruki, Sayaka Matsumoto, Takuya Morikawa, Yusuke Uchiyama, Seiji Kurata, Masayuki Ochi, Yasumasa Ohyagi, Hiroki Shibata
Human Genome Variation.2024;[Epub] CrossRef - Hereditary Ataxias: From Bench to Clinic, Where Do We Stand?
Federica Pilotto, Andrea Del Bondio, Hélène Puccio
Cells.2024; 13(4): 319. CrossRef - Potassium channel‐related epilepsy: Pathogenesis and clinical features
Tong Zhao, Le Wang, Fang Chen
Epilepsia Open.2024;[Epub] CrossRef - Ataxien – Eine aktuelle Übersicht über die weiter wachsende Anzahl möglicher Diagnosen
Andreas Thieme, Dagmar Timmann
Neuroradiologie Scan.2023; 13(01): 63. CrossRef - Zerebellärer Schwindel, was steckt dahinter?
Katharina Feil, Tim W. Rattay, Adedolapo Kamaldeen Adeyemi, Nicolina Goldschagg, Michael Strupp
Nervenheilkunde.2023; 42(01/02): 37. CrossRef - Ataxias: Hereditary, Acquired, and Reversible Etiologies
Chi-Ying R. Lin, Sheng-Han Kuo
Seminars in Neurology.2023; 43(01): 048. CrossRef - SCN2A- Associated Episodic and Persistent Ataxia with Cerebellar Atrophy: A Case Report
Maeve Murray, Jaclyn M. Martindale, Scott I. Otallah
Child Neurology Open.2023; 10: 2329048X2311639. CrossRef - Episodic Ataxias: Primary and Secondary Etiologies, Treatment, and Classification Approaches
Anhar Hassan
Tremor and Other Hyperkinetic Movements.2023;[Epub] CrossRef - Novel Genetic Variants Expand the Functional, Molecular, and Pathological Diversity of KCNA1 Channelopathy
Kelsey Paulhus, Edward Glasscock
International Journal of Molecular Sciences.2023; 24(10): 8826. CrossRef - kcna1a mutant zebrafish model episodic ataxia type 1 (EA1) with epilepsy and show response to first‐line therapy carbamazepine
Deepika Dogra, Paola L. Meza‐Santoscoy, Cezar Gavrilovici, Renata Rehak, Cristiane L. R. de la Hoz, Kingsley Ibhazehiebo, Jong M. Rho, Deborah M. Kurrasch
Epilepsia.2023; 64(8): 2186. CrossRef - A Review of Current and Prospective Treatments for Channelopathies, with a Focus on Gene and Protein Therapy
Monica Sakla, Ulrike Breitinger, Hans-Georg Breitinger, Samar Mansour, Salma Nabil Tammam
Current Pharmaceutical Design.2023; 29(17): 1341. CrossRef - Clinical and VEMPS Characteristics of Benign Recurrent Vertigo With Cochlear Symptoms and Migraine
Xue Zhang, Bo Xu, Yuanquan Li, Yunsheng Wu, Hongbin Yang, Ziming Wu
Ear, Nose & Throat Journal.2023;[Epub] CrossRef - Episodic ataxias
Ján Necpál, Mária Holá, Bibiána Jeleňová, Marcel Árvai
Neurologie pro praxi.2023; 24(4): 253. CrossRef - Case report: Episodic ataxia without ataxia?
Andrea Gaudio, Fabio Gotta, Clarissa Ponti, Francesca Sanguineri, Lucia Trevisan, Alessandro Geroldi, Serena Patrone, Chiara Gemelli, Corrado Cabona, Guja Astrea, Chiara Fiorillo, Stefano Gustincich, Marina Grandis, Paola Mandich
Frontiers in Neurology.2023;[Epub] CrossRef - Genetic Background of Epilepsy and Antiepileptic Treatments
Kinga Borowicz-Reutt, Julia Czernia, Marlena Krawczyk
International Journal of Molecular Sciences.2023; 24(22): 16280. CrossRef - Zerebellärer Schwindel, was steckt dahinter?
Katharina Feil, Tim W. Rattay, Adedolapo Kamaldeen Adeyemi, Nicolina Goldschagg, Michael Leo Strupp
Laryngo-Rhino-Otologie.2023;[Epub] CrossRef - Coincidental occurance of episodic ataxia and multiple sclerosis: a case report and review of the literature
Melike Batum, Ayşın Kısabay Ak, Güldeniz Çetin, Hamide Betül Gerik Çelebi, Sırrı Çam, Hatice Mavioğlu
International Journal of Neuroscience.2022; 132(7): 656. CrossRef - Vestibular impairments in episodic ataxia type 2
Jae-Hwan Choi, Eun Hye Oh, Seo Young Choi, Hyo Jung Kim, Seon Kyung Lee, Jeong Yoon Choi, Ji-Soo Kim, Kwang-Dong Choi
Journal of Neurology.2022; 269(5): 2687. CrossRef - History of Ataxias and Paraplegias with an Annotation on the First Description of Striatonigral Degeneration
José Berciano, José Gazulla, Jon Infante
The Cerebellum.2022; 21(4): 531. CrossRef - The complexities of CACNA1A in clinical neurogenetics
Marina P. Hommersom, Teije H. van Prooije, Maartje Pennings, Meyke I. Schouten, Hans van Bokhoven, Erik-Jan Kamsteeg, Bart P. C. van de Warrenburg
Journal of Neurology.2022; 269(6): 3094. CrossRef - Genetic paroxysmal neurological disorders featuring episodic ataxia and epilepsy
Elisabetta Amadori, Giuditta Pellino, Lalit Bansal, Serena Mazzone, Rikke S. Møller, Guido Rubboli, Pasquale Striano, Angelo Russo
European Journal of Medical Genetics.2022; 65(4): 104450. CrossRef - Ataxien – Eine aktuelle Übersicht über die
weiter wachsende Anzahl möglicher Diagnosen
Andreas Thieme, Dagmar Timmann
Fortschritte der Neurologie · Psychiatrie.2022; 90(05): 233. CrossRef - Episodic ataxias in children and adolescents: Clinical findings and suggested diagnostic criteria
Filipp Maximilian Filippopulos, Lutz Schnabel, Konstanze Dunker, Ralf Strobl, Doreen Huppert
Frontiers in Neurology.2022;[Epub] CrossRef - Vertical Saccadic Slowing in Episodic Ataxia Type 2
Seoyeon Kim, Seondeuk Kim, Seonkyung Lee, Hyo-Jung Kim
Journal of Clinical Neurology.2022; 18(6): 726. CrossRef - Episodic Vestibular Syndrome with Hyperventilation-Induced Downbeat Nystagmus
Eun Hye Oh, Jin-Hong Shin, Jae Wook Cho, Seo Young Choi, Kwang-Dong Choi, Je-Keun Rhee, Jae-Hwan Choi
The Cerebellum.2021; 20(5): 796. CrossRef - Functional and Biochemical Consequences of Disease Variants in Neurotransmitter Transporters: A Special Emphasis on Folding and Trafficking Deficits
Shreyas Bhat, Ali El-Kasaby, Michael Freissmuth, Sonja Sucic
Pharmacology & Therapeutics.2021; 222: 107785. CrossRef - Therapeutic roles of natural remedies in combating hereditary ataxia: A systematic review
Michael Weng Lok Phang, Sze Yuen Lew, Ivy Chung, William Kiong-Seng Lim, Lee Wei Lim, Kah Hui Wong
Chinese Medicine.2021;[Epub] CrossRef - Episodic psychosis, ataxia, motor neuropathy with pyramidal signs (PAMP syndrome) caused by a novel mutation in ADPRHL2 (AHR3)
Hacer Durmus, Elif Mertoğlu, Heinrich Sticht, Serdar Ceylaner, Işın Baral Kulaksızoğlu, Said Hashemolhosseini, Evren Önay Uçar, Yesim Parman
Neurological Sciences.2021; 42(9): 3871. CrossRef - Pediatric Paroxysmal Exercise-Induced Neurological Symptoms: Clinical Spectrum and Diagnostic Algorithm
Federica Rachele Danti, Federica Invernizzi, Isabella Moroni, Barbara Garavaglia, Nardo Nardocci, Giovanna Zorzi
Frontiers in Neurology.2021;[Epub] CrossRef - Paroxysmal Movement Disorders
Susan Harvey, Mary D. King, Kathleen M. Gorman
Frontiers in Neurology.2021;[Epub] CrossRef - Downbeat nystagmus: a clinical review of diagnosis and management
Tu M. Tran, Michael S. Lee, Collin M. McClelland
Current Opinion in Ophthalmology.2021; 32(6): 504. CrossRef - Ataxien – Eine aktuelle Übersicht über die weiter wachsende Anzahl möglicher Diagnosen
Andreas Thieme, Dagmar Timmann
Neurologie up2date.2021; 04(04): 391. CrossRef - Ataxia episódica: 20 años de retraso diagnóstico
I. Muro García, M.E. Toribio-Díaz, S. Quintas
Neurología.2020; 35(7): 500. CrossRef - The expanding spectrum of movement disorders in genetic epilepsies
Apostolos Papandreou, Federica Rachele Danti, Robert Spaull, Vincenzo Leuzzi, Amy Mctague, Manju A Kurian
Developmental Medicine & Child Neurology.2020; 62(2): 178. CrossRef - Episodic Ataxia Secondary to CEP290 Compound Heterozygous Mutations: A Case Report
Moath Hamed, Aakash Shetty, Tara Dzwiniel, Mark Buller, Lotta Koskinen, Oksana Suchowersky
Movement Disorders Clinical Practice.2020; 7(1): 104. CrossRef - Movement Disorders in Genetic Pediatric Ataxias
Simone Gana, Enza Maria Valente
Movement Disorders Clinical Practice.2020; 7(4): 383. CrossRef - Clinical Spectrum of KCNA1 Mutations: New Insights into Episodic Ataxia and Epilepsy Comorbidity
Kelsey Paulhus, Lauren Ammerman, Edward Glasscock
International Journal of Molecular Sciences.2020; 21(8): 2802. CrossRef - Approach to exaggerated startle reflex: a case of hyperekplexia minor
Haris Hakeem, Ramsha Khurshid, Fowzia Siddiqui, Danish Ejaz Bhatti
BMJ Case Reports.2020; 13(4): e232370. CrossRef - Clinical and Genetic Overview of Paroxysmal Movement Disorders and Episodic Ataxias
Giacomo Garone, Alessandro Capuano, Lorena Travaglini, Federica Graziola, Fabrizia Stregapede, Ginevra Zanni, Federico Vigevano, Enrico Bertini, Francesco Nicita
International Journal of Molecular Sciences.2020; 21(10): 3603. CrossRef - Drosophila Glia: Models for Human Neurodevelopmental and Neurodegenerative Disorders
Taejoon Kim, Bokyeong Song, Im-Soon Lee
International Journal of Molecular Sciences.2020; 21(14): 4859. CrossRef - The Genetic Relationship between Paroxysmal Movement Disorders and Epilepsy
Hyunji Ahn, Tae-Sung Ko
Annals of Child Neurology.2020; 28(3): 76. CrossRef - Episodic ataxia: a 20-year diagnostic delay
I. Muro García, M.E. Toribio-Díaz, S. Quintas
Neurología (English Edition).2020; 35(7): 500. CrossRef - TRPM7 as a Candidate Gene for Vestibular Migraine
Eun Hye Oh, Jin-Hong Shin, Jae Wook Cho, Seo-Young Choi, Kwang-Dong Choi, Jae-Hwan Choi
Frontiers in Neurology.2020;[Epub] CrossRef - Ictal downbeat nystagmus in Ménière disease
Sun-Uk Lee, Hyo-Jung Kim, Jeong-Yoon Choi, Ji-Soo Kim
Neurology.2020;[Epub] CrossRef - Involvement of the Peripheral Nervous System in Episodic Ataxias
Wojciech Koźmiński, Joanna Pera
Biomedicines.2020; 8(11): 448. CrossRef - Acetazolamide-Responsive Episodic Ataxia Linked to Novel Splice Site Variant in FGF14 Gene
M. Schesny, F. Joncourt, Alexander A. Tarnutzer
The Cerebellum.2019; 18(3): 649. CrossRef - Paroxysmal movement disorders: Recent advances and proposal of a classification system
Xiao-jin Zhang, Zhe-yu Xu, Yun-cheng Wu, Eng-King Tan
Parkinsonism & Related Disorders.2019; 59: 131. CrossRef - Acute cerebellar ataxia: differential diagnosis and clinical approach
José Luiz Pedroso, Thiago Cardoso Vale, Pedro Braga-Neto, Lívia Almeida Dutra, Marcondes Cavalcante França Jr, Hélio A. G. Teive, Orlando G. P. Barsottini
Arquivos de Neuro-Psiquiatria.2019; 77(3): 184. CrossRef - Acute vestibular asymmetry disorder: a new disease entity in acute vestibular syndrome?
Se A. Lee, Eek-Sung Lee, Bo Gyung Kim, Tae-Kyeong Lee, Ki-Bum Sung, Kyurin Hwang, Jong Dae Lee
Acta Oto-Laryngologica.2019; 139(6): 511. CrossRef - Paroxysmal Movement Disorders: Recent Advances
Zheyu Xu, Che-Kang Lim, Louis C. S. Tan, Eng-King Tan
Current Neurology and Neuroscience Reports.2019;[Epub] CrossRef - Paroxysmal movement disorders – practical update on diagnosis and management
Claudio M. De Gusmao, Laura Silveira-Moriyama
Expert Review of Neurotherapeutics.2019; 19(9): 807. CrossRef - Clinical manifestations of episodic ataxia type 5
Miguel González Sánchez, Silvia Izquierdo, Sara Álvarez, Rosa Eva Bautista Alonso, José Berciano, José Gazulla
Neurology Clinical Practice.2019; 9(6): 503. CrossRef - Pearls & Oy-sters: Paroxysmal dysarthria-ataxia syndrome
Annalisa Gessani, Francesco Cavallieri, Carla Budriesi, Elisabetta Zucchi, Marcella Malagoli, Sara Contardi, Maria Teresa Mascia, Giada Giovannini, Jessica Mandrioli
Neurology.2019;[Epub] CrossRef - Consensus Paper: Neurophysiological Assessments of Ataxias in Daily Practice
W. Ilg, M. Branscheidt, A. Butala, P. Celnik, L. de Paola, F. B. Horak, L. Schöls, H. A. G. Teive, A. P. Vogel, D. S. Zee, D. Timmann
The Cerebellum.2018; 17(5): 628. CrossRef - The expanding spectrum of paroxysmal movement disorders: update from clinical features to therapeutics
Eavan M. McGovern, Emmanuel Roze, Timothy J. Counihan
Current Opinion in Neurology.2018; 31(4): 491. CrossRef - Benign paroxysmal torticollis of infancy does not lead to neurological sequelae
Annika Danielsson, Britt‐Marie Anderlid, Tommy Stödberg, Kristina Lagerstedt‐Robinson, Eva Klackenberg Arrhenius, Kristina Tedroff
Developmental Medicine & Child Neurology.2018; 60(12): 1251. CrossRef - Epidemiology of inherited cerebellar ataxias and challenges in clinical research
Federica Pilotto, Smita Saxena
Clinical and Translational Neuroscience.2018; 2(2): 2514183X1878525. CrossRef - Ataxia in children: early recognition and clinical evaluation
Piero Pavone, Andrea D. Praticò, Vito Pavone, Riccardo Lubrano, Raffaele Falsaperla, Renata Rizzo, Martino Ruggieri
Italian Journal of Pediatrics.2017;[Epub] CrossRef - Episodic Ataxia Type 1 (K‐channelopathy) Manifesting as Paroxysmal Nonkinesogenic Dyskinesia: Expanding the Phenotype
Kallol K. Set, Debabrata Ghosh, A.H.M. Huq, Aimee F. Luat
Movement Disorders Clinical Practice.2017; 4(5): 784. CrossRef - Genetic Variants Associated with Episodic Ataxia in Korea
Kwang-Dong Choi, Ji-Soo Kim, Hyo-Jung Kim, Ileok Jung, Seong-Hae Jeong, Seung-Han Lee, Dong Uk Kim, Sang-Ho Kim, Seo Young Choi, Jin-Hong Shin, Dae-Seong Kim, Kyung-Pil Park, Hyang-Sook Kim, Jae-Hwan Choi
Scientific Reports.2017;[Epub] CrossRef - Episodic dizziness and vertigo
Štefan Sivák
Neurologie pro praxi.2017; 18(3): 156. CrossRef
 E-submission
E-submission

 PubReader
PubReader ePub Link
ePub Link Cite
Cite
