Episodic Ataxias: Clinical and Genetic Features
Article information
Abstract
Episodic ataxia (EA) is a clinically heterogeneous group of disorders that are characterized by recurrent spells of truncal ataxia and incoordination lasting minutes to hours. Most have an autosomal dominant inheritance pattern. To date, 8 subtypes have been defined according to clinical and genetic characteristics, and five genes are known to be linked to EAs. Both EA1 and EA2, which are caused by mutations in KCNA1 and CACNA1A, account for the majority of EA, but many patients with no identified mutations still exhibit EA-like clinical features. Furthermore, genetically confirmed EAs have mostly been identified in Caucasian families. In this article, we review the current knowledge on the clinical and genetic characteristics of EAs. Additionally, we summarize the phenotypic features of the genetically confirmed EA2 families in Korea.
INTRODUCTION
Episodic ataxia (EA) is a clinically heterogeneous group of disorders that are characterized by recurrent spells of truncal ataxia and incoordination [1,2]. The incidence is likely to be less than 1/100,000, but it may be underestimated due to demanding genetic tests and unidentified causative genes. Most have an autosomal dominant inheritance pattern, although some sporadic cases have been reported. There are several subtypes of EA, and these subtypes are defined according to clinical features and genetic characterizations. However, clinical and genetic heterogeneity is not uncommon (Table 1). Only EA1 and EA2 have been reported in multiple families of different ethnicity. They are caused by mutations involving the potassium and calcium channel genes, KCNA1 and CACNA1A. Mutations in CACNB4 and SLC1A3 are known to cause EA5 and EA6, respectively, but have been reported in only one or two families [3-5]. Recently, by whole-exome sequencing, UBR4 was found to be associated with EA [6].

Genetic and clinical summary of episodic ataxia (EA)
In Korea, genetically confirmed EA has rarely been reported, probably due to clinical heterogeneity and the limited availability of commercial genetic tests [7-10]. However, recent advances in molecular genetics have led to the increased identification of candidate variants and new mutations associated with EAs.
This review introduces the updates on the clinical and genetic features of EAs and the phenotypes of Korean EA patients that have been reported in the literature.
CLINICAL FEATURES
The key clinical feature of EAs is the discrete attacks of incoordination, with a clear onset and resolution of symptoms [1,2]. However, some patients can also have progressive ataxia, which makes it difficult to distinguish EA with progressive features from progressive ataxia with intermittent exacerbation. Furthermore, the clinical features of EA subtypes overlap each other. Among the various subtypes, only EA1 and EA2 have the characteristic interictal findings and well-defined clinical features.
EA1
EA1 is characterized by brief episodes of ataxia with constant interictal myokymia [1,2,11,12]. The onset is typically during early childhood. The core features are imbalance, incoordination and slurred speech and are triggered by physical and emotional stress, startle response, or abrupt movements. The typical duration of attacks lasts seconds to minutes, but prolonged attacks lasting hours or days have also been described [13]. Furthermore, a recent large prospective study revealed that at least one-fifth of EA1 patients developed permanent cerebellar symptoms and signs [11]. Myokymia may be clinically evident or only detectable by electromyography. Neuromyotonia is also apparent in most patients, to a varying degree, and is characterized by muscle stiffness, twitching, flickering, and muscle hypertrophy. The presence of myokymia or neuromyotonia suggests the involvement of the peripheral nervous system. Additional symptoms include distal weakness, tremors, choreoathetosis, spinning sensation, and cognitive dysfunctions.
EA2
EA2 is the most common subtype of EA. The episodes are characterized by recurrent ataxia, slurred speech for several hours, and interictal nystagmus. The onset is typically early in life, but patients with an onset during the sixth decade have also been reported [8,14,15]. Vertigo and fluctuating general weakness are common. Additional features include diplopia, tinnitus, seizure, dystonia, and cognitive impairment. Some patients initially present with a paroxysmal tonic upward gaze lasting a few minutes to one hour during infancy, and the patients then gradually develop cerebellar symptoms as the tonic upward gaze disappears [16,17]. Similarly, in EA1, the attacks are commonly triggered by exercise, or physical or emotional stress. A recent study described that severe postural instability with pure downbeat nystagmus was provoked by intense exercise in a patient with EA2 [8]. Between the attacks, patients may be free of symptoms but can present mildly progressive baseline ataxia. Most patients also show various interictal nystagmus, such as gaze-evoked nystagmus (GEN), rebound nystagmus, or primary position downbeat nystagmus. Acetazolamide can prevent the attacks by reducing arterial pH, but it may cause kidney stones. Alternatively, aminopyridine, a nonselective blocker of voltage-gated potassium channels, reduces the frequency of attacks and improves the quality of life in patients with EA2 [18].
Other EA subtypes
Other EA subtypes have only been reported in one or two families.
EA3 was described in a single large Canadian family [19]. They were characterized by recurrent episodes of truncal ataxia, vertigo, and tinnitus, usually lasting less than 30 minutes. Other features included interictal myokymia, headache, visual blurring, diplopia, and weakness. The presence of vertigo and tinnitus and the absence of interictal nystagmus and shorter episodes distinguish EA3 from EA1 and EA2.
EA4, which is also called periodic vestibulocerebellar ataxia, was described in two North Carolina kindreds, suggesting a single common founder [20,21]. They were characterized by ataxia, vertigo, GEN, and defective smooth pursuit. The age of onset ranged from the third to sixth decades. The attacks typically last hours and do not response to acetazolamide.
EA5, which is caused by a mutation in CACNB4, was reported in a French Canadian family [3]. The clinical features were similar to those of EA2, including the duration of attacks, interictal nystagmus, and response to acetazolamide. The only obvious difference is that its onset is later than that of EA2.
EA6 is caused by a mutation in SLC1A3 and was identified in two unrelated individuals [4,5]. Their clinical phenotypes were different from each other. One sporadic child presented with a severe form of EA with seizure, migraine, and alternating hemiplegia, which was triggered by febrile illness [4]. The other Dutch family showed typical EA2-like symptoms, such as episodes with a duration of several hours, interictal nystagmus, and a positive response to acetazolamide [5].
EA7 was reported in a 4-generation family, in which 7 members had EA [22]. The clinical features were similar to those of EA2, except without interictal findings.
Recently, the Online Mendelian Inheritance of Man database registered a large 3-generation Irish family with EA as EA8 [6]. The attacks were characterized by unsteadiness, general weakness, and slurred speech. Additional features included twitching around the eyes, nystagmus, myokymia, and persistent intention tremor. An interesting finding was the response to clonazepam, instead of acetazolamide, which was consistent within the affected individuals.
GENETIC CHARACTERISTICS
There are at least 8 loci for EA, 5 of which are known genes (Table 1). All of the identified genes, except UBR4, encode ion channel proteins located on the neuronal or glial membrane and play important roles in excitatory neurotransmission.
EA1: KCNA1
KCNA1, which is located on chromosome 12p13, is the gene responsible for EA1 [1,2,11,12]. It encodes the voltage-gated potassium channel, Kv1.1, which is expressed highly in basket cells and interneurons forming the GABAergic synapses on Purkinje cells. Kv1.1 plays an important role in the repolarization phase of presynaptic action potentials that affect the inhibitory inputs to the Purkinje cells. Thus, KCNA1 mutations result in the hyperexcitability of the presynaptic basket cells and the excessive release of the neurotransmitter, GABA, which can inhibit the generation of action potentials in the Purkinje cells. As a consequence, the inhibitory output of the cerebellum may be markedly reduced, thus producing the cerebellar symptoms observed in EA1 patients.
To date, 30 mutations have been identified in EA1 individuals, and the majority are missense point mutations [1,11,12]. Four different mutations have been described on the highly conserved threonine residue at codon 226 (T226M/A/R/K), which is located in the second transmembrane segment, but the phenotypic characteristics are diverse. This can be explained by the extent of functional impairment related to the potassium channel, depending on the type of KCNA1 mutation. In other words, all mutations result in loss-of-function effects on the outward K+ flux by altering channel expression and gating, but the effect of each mutation is highly heterogeneous. Furthermore, due to wide interfamilial and intrafamilial phenotypic variability, genotype-phenotype correlations are difficult to establish [11].
EA2: CACNA1A
EA2 is caused by mutations involving CACNA1A on chromosome 19p13, which encodes Cav2.1, the α1 subunit of the P/Q-type voltage-gated calcium channel [1,2]. This channel is widely expressed in the Purkinje and granule cells of the cerebellum; it mediates calcium entry into the cells and regulates the precision of pacemaking. Thus, CACNA1A mutations lead to a decrease in Ca2+ entry through Cav2.1 and the irregular firing of the Purkinje cells, which contributes to EA2 symptoms.
Over 60 different heterozygous point mutations have been described in EA2 patients. Most mutations disrupt the open reading frame and subsequently lead to a premature stop due to nonsense mutation or defects in splice sites. However, some missense mutations have also been reported. In some EA2 patients, the direct sequencing of CACNA1A did not identify any point mutation, but the use of multiplex ligation-dependent probe amplification demonstrated large genomic deletions or duplications [23,24]. Although missense mutations typically involve the pore loop region of the protein, CACNA1A mutations occur throughout the entire gene, without any consistent hot spots.
CACNA1A is also the gene responsible for familial hemiplegic migraine type 1 (FHM1) and spinocerebellar ataxia type 6 (SCA6). However, in FHM1 and SCA6, increased Ca2+ influx through the mutant channel causes aberrant neurotransmitter release and excitotoxicity [1]. Nevertheless, there is clinical overlap among EA2, FHM1, and SCA6. Many patients with FHM1 show cerebellar symptoms and signs, whereas over half of the EA2 patients have migraines that meet the International Headache Society criteria. Some patients with SCA6 also present with fluctuating ataxia similar to EA2.
Other EAs
In addition to KCNA1 and CACNA1A, there are 3 other EA genes.
EA5 results from mutations involving CACNB4, which encodes the calcium channel β4 subunit and is located on chromosome 2q22-23 [3]. This subunit interacts directly with the C-terminus of α1 subunit (Cav2.1) and contributes to the modulation of calcium current amplitude, voltage dependence, and kinetics of activation and inactivation. A missense mutation C104F has been identified in an EA5 family, but the functional alterations of channel kinetics were subtle. CACNB4 mutations have also been reported in a family with generalized epilepsy (C104F) and juvenile myoclonic epilepsy (R482X).
EA6 is caused by mutations in SLC1A3 on chromosome 5p, which encodes excitatory amino acid transporter 1 (EAAT1), a glial glutamate transporter [4,5]. EAAT1 is present in the brainstem and cerebellum and is responsible for glutamate uptake in the synapses. Thus, SLC1A3 mutation leads to the excessive extracellular accumulation of glutamate and neurotoxic insults. Only 2 mutations, P290R and C186S, have been reported in the literature, and the mutant EAAT1 has been shown to result in decreased glutamate uptake in cell culture assays.
Recently, the missense variant, R5091H, in UBR4, which is located on chromosome 1p36.13, was found in a large Irish family with autosomal dominant EA8 by whole-exome sequencing [6]. However, its contribution to EA8 has not been confirmed by functional studies. UBR4 is known to interact with calmodulin, a Ca2+ regulating protein in the cytoplasm, and may thus be involved in the regulation of neuronal excitability.
Although recent advances in next-generation sequencing techniques have contributed to the easy identification of pathogenic mutations, many patients with typical clinical features of EA do not have any mutations in the known EA genes. This suggests a genetic heterogeneity of EA and the presence of additional causative EA genes. EA3 and EA7 are linked to chromosome 1q42 and 19q13, respectively, but no candidate gene has been confirmed [19,22]. In EA4, linkage analysis ruled out EA1 and EA2 loci, but to date, no genome-wide scan has been reported [21].
Many genes coding the ion channels, transporters, or synaptic proteins play crucial roles in regulating neural excitability in the central nervous system. Mutations in these genes can cause various paroxysmal neurological symptoms, and several overlapping syndromes have been described, of which EA has been reported as one of various phenotypes (Table 2). For example, mutations in SCN2A, encoding the voltage-gated Na+ channel, Nav 1.2, cause benign familial neonatal-infantile seizures but can also contribute to later onset EA [25,26]. Other genes associated with EA include ATP1A3 [27], NALCN [28], DARS2 [29], SLC2A1 [30], FGF14 [31], and PRRT2 [32], and they have been reported to cause epilepsy and paroxysmal dyskinesia. Thus, extensive searches for these genes with whole-exome sequencing may help define novel candidate genes and identify new mutations associated with EA.

Other genes associated with episodic ataxia (EA)
EAS IN KOREA
To date, four Korean EA2 families have been reported in the literature (Table 3, Figure 1) [7-10]. Two splice site mutations, one nonsense mutation and one missense mutation involving CACNA1A were identified, and one of them was previously reported as a pathogenic mutation in a Caucasian EA2 patient [33]. The clinical features of the Korean patients were similar to those of Caucasian EA2 patients, including recurrent episodes of ataxia or vertigo lasting hours, interictal nystagmus and a good response to acetazolamide. However, each family had an intriguing feature. In the first family with a deletion mutation (c.2042_2043delAG), the symptoms were provoked by a change in body temperature, such as heat or high fever [7]. This suggests that calcium channel function may have a strong dependence on body temperature. The second family with a nonsense mutation (c.3832C>T) presented with recurrent ataxia and vertigo that were frequently triggered by exercise, and one member developed pure downbeat nystagmus and severe postural imbalance after exercise [8]. Ictal downbeat nystagmus may be ascribed to changes in the cerebellar pH homeostasis precipitated by hyperventilation during the exercise. The third family with a splice site mutation (c.4392-1G>C) showed a decrease in the age of onset, possibly due to genetic anticipation, in three succeeding generation [9]. Anticipation mostly occurs in neurodegenerative diseases that are characterized by an expansion of unstable trinucleotide repeats, but it has not been reported in EA2. In the last family with another splice site mutation (c.4953+1G>A), one patient showed transient upbeat nystagmus on resuming primary eye position after lateral gazes that had induced GEN and downbeat nystagmus [10]. This phenomenon can be explained by a shifting null in the vertical planes as a result of an adaptation to the downbeat nystagmus that developed during lateral gaze.

Literature reports of Korean families with episodic ataxia type 2
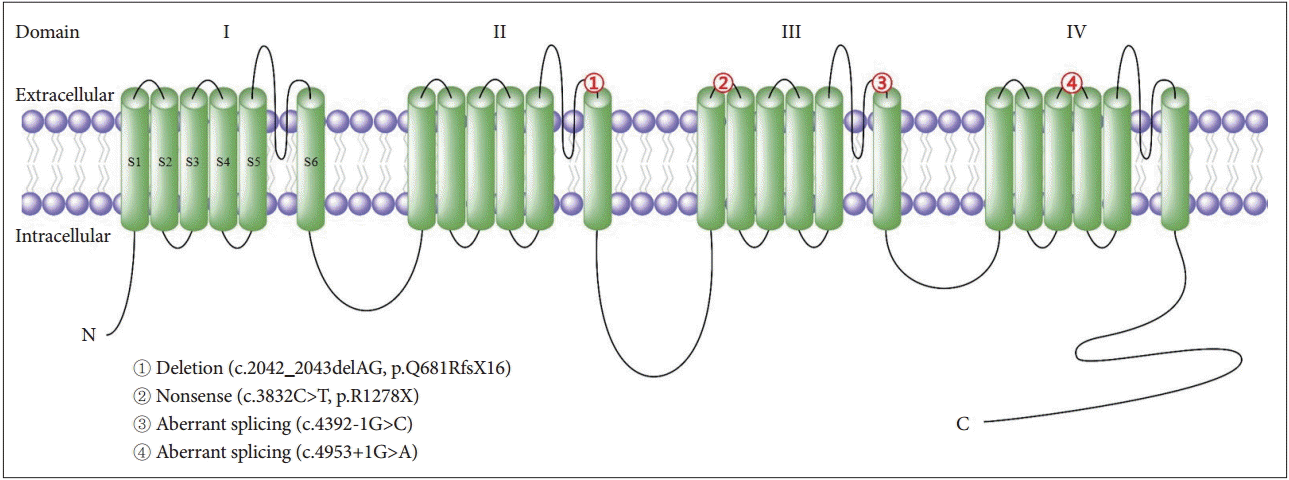
Mutations of the α1 subunit of the P/Q-type voltage-gated calcium channel in Korean patients with episodic ataxia type 2. The protein contains four homologous domains (I–IV), each with six transmembrane segments (S1–S6). The numbers in the symbol correspond to the mutations listed in Table 3.
Although only four Korean EA2 families have been reported in the literature and other subtypes have not yet been identified, this does not mean that the frequency of EA is lower in the Korean population compared to Caucasian populations. Although there are many clinically diagnosed EA patients without genetic confirmations in multiple centers, the limited availability of genetic tests make it difficult to establish a genetic confirmation. As whole-exome sequencing becomes more affordable, the identification of new mutations associated with EAs will increase in the future.
Notes
Conflicts of Interest
The authors have no financial conflicts of interest.
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education (NRF-2015R1D1A1A01060559).