 E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 10(2); 2017 > Article
-
Letter to the editor
Beyond the Classic Segawa Disease, GCH1-Associated Neurodegenerative Parkinsonism: Practical Considerations for Physicians - Jirat Chenbhanich1,2, Jirada Sringean1, Roongroj Bhidayasiri1,3
-
Journal of Movement Disorders 2017;10(2):102-104.
DOI: https://doi.org/10.14802/jmd.17009
Published online: April 18, 2017
1Chulalongkorn Center of Excellence for Parkinson’s Disease & Related Disorders, King Chulalongkorn Memorial Hospital, Thai Red Cross Society, Bangkok, Thailand
2Department of Internal Medicine, MetroWest Medical Center, Framingham, MA, USA
3Department of Neurology, Juntendo University Hospital, Tokyo, Japan
- Corresponding author: Roongroj Bhidayasiri, MD, FRCP, FRCPI, Chulalongkorn Center of Excellence for Parkinson’s Disease & Related Disorders, King Chulalongkorn Memorial Hospital, 1873 Rama 4 Road, Bangkok 10330, Thailand / Tel: +66-2-256-4000 ext. 70701 / Fax: +66-2-256-4630 / E-mail: rbh@chulapd.org
• Received: February 3, 2017 • Revised: February 22, 2017 • Accepted: February 24, 2017
Copyright © 2017 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- We would like to express our gratitude to Dr. Objoon Trachoo, Division of Genetics, Department of Medicine, Ramathibodi Hospital for genetic analysis and Dr. Supatporn Tepmongkol, Division of Nuclear Medicine, Department of Radiology, Faculty of Medicine, Chulalongkorn University for the interpretation of [18F]DOPA PET imaging.
Acknowledgments
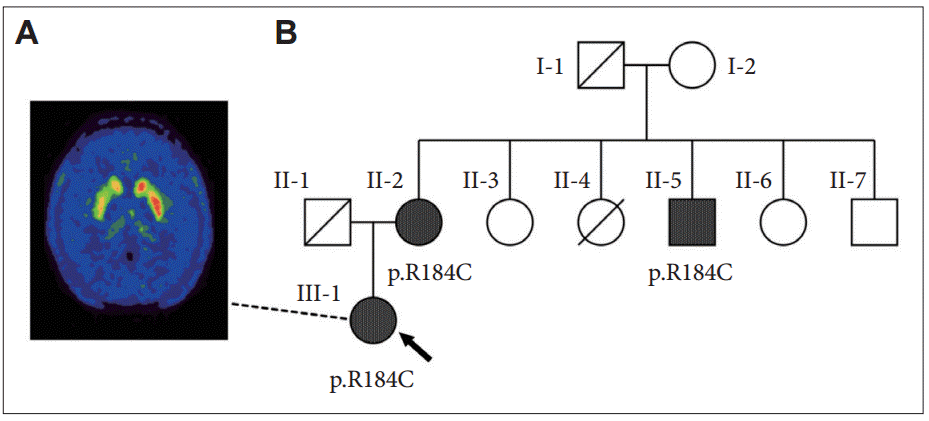
Figure 1.Patientʼs [18F]DOPA-PET imaging and her pedigree. A: [18F]DOPAPET of our patient (III-1) showing decreased [18F]DOPA uptake in the right caudate and putamen, indicating a degenerative etiology. B: Pedigree showing two additional individuals with the same pathogenic heterozygous GCH1 variant (R184C): the patient (arrow), her 73-year-old mother (II-2) who complained of slowing gait beginning one year ago, and her 60-year-old uncle (II-5) who has a history of alcoholism and has had ataxia for two years. Individual II-4 died from perinatal pneumonia. Diagnosed with Parkinsonism at 80 years old, her grandfather (I-1) had received levodopa for two years before dying of aspiration pneumonia. None of the family members had dystonia. [18F]DOPA-PET: 18F-dihydroxyphenylalanine positron emission tomography.
- 1. Wijemanne S, Jankovic J. Dopa-responsive dystonia--clinical and genetic heterogeneity. Nat Rev Neurol 2015;11:414–424.ArticlePubMedPDF
- 2. Mencacci NE, Isaias IU, Reich MM, Ganos C, Plagnol V, Polke JM, et al. Parkinson’s disease in GTP cyclohydrolase 1 mutation carriers. Brain 2014;137(Pt 9):2480–2492.ArticlePubMedPMC
- 3. Bandmann O, Valente EM, Holmans P, Surtees RA, Walters JH, Wevers RA, et al. Dopa-responsive dystonia: a clinical and molecular genetic study. Ann Neurol 1998;44:649–656.ArticlePubMed
- 4. Hagenah J, Saunders-Pullman R, Hedrich K, Kabakci K, Habermann K, Wiegers K, et al. High mutation rate in dopa-responsive dystonia: detection with comprehensive GCHI screening. Neurology 2005;64:908–911.ArticlePubMed
- 5. Kikuchi A, Takeda A, Fujihara K, Kimpara T, Shiga Y, Tanji H, et al. Arg(184)His mutant GTP cyclohydrolase I, causing recessive hyperphenylalaninemia, is responsible for doparesponsive dystonia with parkinsonism: a case report. Mov Disord 2004;19:590–593.ArticlePubMed
- 6. Trender-Gerhard I, Sweeney MG, Schwingenschuh P, Mir P, Edwards MJ, Gerhard A, et al. Autosomal-dominant GTPCH1-deficient DRD: clinical characteristics and long-term outcome of 34 patients. J Neurol Neurosurg Psychiatry 2009;80:839–845.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
Citations to this article as recorded by 
- Comprehensive Analysis of Familial Parkinsonism Genes in Rapid‐Eye‐Movement Sleep Behavior Disorder
Kheireddin Mufti, Uladzislau Rudakou, Eric Yu, Lynne Krohn, Jennifer A. Ruskey, Farnaz Asayesh, Sandra B. Laurent, Dan Spiegelman, Isabelle Arnulf, Michele T.M. Hu, Jacques Y. Montplaisir, Jean‐François Gagnon, Alex Desautels, Yves Dauvilliers, Gian Luigi
Movement Disorders.2021; 36(1): 235. CrossRef - Differences in Drug Pharmacokinetics and Motor Fluctuation in DYT-GCH1
Gerard Saranza, Anthony E. Lang
Canadian Journal of Neurological Sciences / Journal Canadien des Sciences Neurologiques.2021; 48(5): 734. CrossRef - A Compound Heterozygote for GCH1 Mutation Represents a Case of Atypical Dopa-Responsive Dystonia
Subhajit Giri, Tufan Naiya, Shubhrajit Roy, Gautami Das, Gurusidheshwar M. Wali, Shyamal Kumar Das, Kunal Ray, Jharna Ray
Journal of Molecular Neuroscience.2019; 68(2): 214. CrossRef - Common and rare GCH1 variants are associated with Parkinson disease
Uladzislau Rudakou, Bouchra Ouled Amar Bencheikh, Jennifer A. Ruskey, Lynne Krohn, Sandra B. Laurent, Dan Spiegelman, Christopher Liong, Stanley Fahn, Cheryl Waters, Oury Monchi, Edward A. Fon, Yves Dauvilliers, Roy N. Alcalay, Nicolas Dupré, Ziv Gan-Or
Neurobiology of Aging.2018;[Epub] CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite
