ABSTRACT
- Mutations in presenilin 1 (PSEN1) are the most common cause of autosomal dominant Alzheimer’s disease. Here, we report a Canadian-Vietnamese family carrying a PSEN1 p.Met233Val mutation with an exceptionally early and severe presentation that includes a wide range of atypical symptoms, including prominent ataxia, Parkinsonism, spasticity, dystonia, action tremor, myoclonus, bulbar symptoms, seizures, hallucinations and behavioral changes. Whole-exome sequencing (WES) was performed on the affected proband after many assessments over several years proved diagnostically inconclusive. The results were analyzed using the AnnEx “Annotated Exomes” browser (http://annex.can.ubc.ca), a web-based platform that facilitates WES variant annotation and interpretation. High-throughput sequencing can be especially informative for complex neurological disorders, and WES warrants consideration as a first-line clinical test. Data analyses facilitated by web-based bioinformatics tools have great potential for novel insight, although confirmatory, diagnostically accredited Sanger sequencing is recommended prior to reporting.
-
Keywords: Neurodegeneration; exome; bioinformatics; Alzheimer; mutation
Mutations in presenilin 1 (PSEN1) are the most common cause of autosomal dominant Alzheimer’s disease (AD), with over 215 different mutations reported so far (http://www.molgen.vib-ua.be/ADMutations) [1]. PSEN1 heterozygotes have varied clinical presentations, although a correlation between the mutation site and the phenotype is noted [2]. Here, we present a Canadian-Vietnamese family in which affected subjects have a PSEN1 p.Met233Val mutation. The proband has an unusually low age of onset and atypical neurological features.
CASE REPORT
- The affected proband (III-2) (Figure 1) had a history of substance abuse (alcohol, ecstasy, crack cocaine and marijuana, but not intravenous drugs) from age 16 to 22. At age 22, she developed gradually progressive memory and global cognitive problems, and her Montréal cognitive assessment (MoCA) at age 26 was 15/30. At age 27, a neurological exam revealed gait and appendicular ataxia, dystonia, slow speech and a shuffling gait. She had auditory hallucinations and exhibited verbally aggressive behaviors. At age 29, she developed generalized tonic-clonic and myoclonic seizures and urinary incontinence. Examination demonstrated bradykinesia, rigidity, spasticity and action tremor. By age 30, she had severe dysphagia and was immobile, incontinent and mute. She had frequent myoclonic jerks and pronounced rigidity, and she was unable to follow commands.
- Cerebrospinal fluid collected at age 27 was normal for cells and protein, oligoclonal bands were negative, and both the 14-3-3 protein assay and the quaking-induced conversion assay (carried out when the patient was 31) to test for prion disease were negative. At the age of 28, an MRI revealed mild to moderate, generalized, predominantly supratentorial atrophy. Testing was normal for human immunodeficiency virus, syphilis, hepatitis B, hepatitis C, N-methyl-D-aspartate receptor antibodies and paraneoplastic antibodies, vitamin E and B12, copper collected over 24 hours in urine, ceruloplasmin, Kayser-Fleischer rings, creatine kinase, liver function and renal function. Her pelvic ultrasound was normal. Genetic testing for spinocerebellar ataxia types 1, 2, 3, 6, and 7; Huntington’s disease; Friedreich’s ataxia; and dentato-rubral pallidoluysian atrophy was negative. Electroencephalography (EEG) at age 28 revealed a very disorganized background and spike-wave activity. An EEG at age 31 showed moderate slowing, one short myoclonic seizure and several myocloni without EEG equivalent. There is no history of any neurological issues on the maternal side. Medical records for the proband’s father (II-3) describe hallucinations that began at age 15, and by age 30, he was drug and alcohol dependent. At age 32, disorientation, slurred and incoherent speech, gait disturbance, paranoia and severe aggression were noted. He was admitted to a long-term psychiatric institution. At age 36, severe ataxia, wandering, urinary and fecal incontinence, inappropriate laughing and auditory and possibly visual hallucinations were described. His Mini Mental Status Examination was 11/30. He lost his ability to walk, became mute and had generalized tonic-clonic seizures, and he died at age 40.
- Anecdotally, the paternal grandfather (I-1) died at the age of 45. His death was attributed to alcohol abuse. A paternal uncle (II-2) is also described to have had memory and balance issues that were possibly related to alcohol abuse. The affected proband’s older brother (III-1) developed a progressively unsteady gait and progressive cognitive problems at the age of 15. At age 32, his exam showed appendicular and gait ataxia with falls, spastic paraparesis, rigidity and mild dysarthria. He scored 16/30 on the MoCA. At age 34, he developed dysphagia, myoclonus, and behavioral changes, including verbal aggression, apathy and repetitive behaviors. He had episodes suggestive of absence seizures, and his EEG was markedly abnormal with fragments of spike-and slow-wave discharges.
- The affected proband underwent numerous laboratory and imaging studies and several single gene tests, which led to the identification of a prion protein (PRNP) mutation (NM_000311: c.290G > A, p.Ser97Asn) previously described in a subject presenting with sporadic AD [3]. The unresolved clinical presentation and the finding of a PRNP variant were suggestive of a transmissible spongiform encephalopathy. Infection control precautions were implemented making further interventions and laboratory testing challenging. However, we determined the PRNP mutation was inherited from the unaffected mother and was not shared by the affected brother. The lack of disease segregation and absence of putative damage to protein structure [Combined Annotation Dependent Depletion (CADD) = 0.001] [4] indicates that the mutation is unlikely pathogenic.
- At age 31, high-throughput whole-exome sequencing (WES) was performed for the proband using the Ion ProtonTM System according to the manufacturer’s recommendations (Thermo Fisher Scientific, Carlsbad, CA, USA). Exonic regions were captured with the Ion AmpliSeq Exome Kit (57.7 Mb; Thermo Fisher Scientific). Sequence reads were aligned against the human reference genome (hg19). The results were analyzed using the AnnEx “Annotated Exomes” browser (http://annex.can.ubc.ca), a contemporary web-based compendium of publicly available genetic resources to facilitate WES variant annotation. AnnEx includes expert-curated panels of genes linked to specific neurological disorders. Subsequent filtering was restricted to the patient’s predominant symptoms and genes linked to ‘dementia’ (59 genes), ‘Parkinsonism’ (36 genes) and ‘ataxia’ (148 genes). Analysis of the overlapping set of 229 genes revealed 11 variants, of which two heterozygous mutations are of particular clinical relevance: 1) PSEN1 (NM_000021: c.697A > G, p.Met233Val), presumed structurally deleterious (CADD score = 25.7), not observed in control subjects [Exome annotation consortium (ExAC) allele frequency = 0] (http://exac.broadinstitute.org/) and referenced pathogenic in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and 2) PSEN2 (NM_ 012486:c.100G > A:p.Gly34Ser), which is modestly damaging to protein structure (CADD score = 18.6), rarely observed in control subjects (ExAC allele frequency = 0.0005) and without reference in ClinVar. The PRNP (NM_ 183079:c.290G > A, p.Ser97Asp) variant was observed and confirmed prior testing. All three mutations were validated by Sanger sequencing, but only PSEN1 p.Met233Val was shown to segregate with disease (Figure 1).
DISCUSSION
- This family has an exceptionally early and severe presentation that includes a wide range of atypical symptoms. The age of onset was in the second and third decade, whereas individuals with AD due to PSEN1 mutations typically present between 40 and 50 years [5]. Drug abuse may have been a contributor to and/or a consequence of the disease, but symptoms clearly progressed for years after the cessation of drug consumption. Of note, the affected brother never used recreational drugs, but behavioral and delusional presentations are documented in patients with PSEN1 mutations [6]. The father of the affected proband did not receive a diagnosis during his lifetime. No test results were diagnostically conclusive before the WES analysis of the proband despite numerous prior studies performed over many years.
-
PSEN1 p.Met233Val has been described in another family with an early-onset (third and fourth decade) and rapidly progressive disease with extrapyramidal symptoms and seizures. Pathological examination showed amyloid plaques, neurofibrillary tangles and extensive Lewy body formation [7]. PSEN1 p.Met233Val also occurs at the same residue as three other substitutions: p.Met233Thr [8], p.Met233Leu [6], and p.Met233Ile [9], all of which are associated with early-onset, aggressive, familial AD with a similarly broad range of atypical symptoms, including executive dysfunction and behavioral changes, dysarthria, apraxia, dystonia, myoclonus, seizures and pyramidal signs (Babinski) [8,10]. Nevertheless, in such families, disease-modifying interventions targeting PSEN1 and the amyloid pathway hold greatest promise. Within a family, patients may have variable onset and phenotypes even when an autosomal dominant, monogenic Mendelian pattern of inheritance is observed because penetrance/expressivity is influenced by other genetic, environmental and stochastic factors. Sanger sequencing and segregation analysis shows PSEN1 p.Met233Val is necessary and sufficient for disease (etiologic) (Figure 1). It is unclear whether the rare coding variability observed in PSEN2 p.Gly34Ser and PRNP p.Ser97Asp contributes to disease ontology. However, PSEN1, PSEN2, and PRNP proteins function in the same biological network and the proband had an especially early and rapid demise. Functional studies may provide more insight in this regard.
- In complex neurological disorders, high-throughput sequencing can be especially informative. Indeed, WES should be considered as a first-line clinical test given the ease of genome-wide assessment, the high diagnostic yield, the accuracy of mutation detection, and the relatively low cost of DNA extraction from a mouthwash or blood sample. Web-based bioinformatics tools now simplify and accelerate data review relevant to symptoms. Genetic mutations have prognostic value and increasingly have treatment implications, and, with informed consent, may enable longitudinal re-analyses and discovery. This is especially important with disease development and when symptoms present insidiously. Nevertheless, prior to reporting, we recommend any WES findings are confirmed as pathogenic by diagnostically accredited Sanger sequencing.
Notes
-
Conflicts of Interest
The authors have no financial conflicts of interest.
Acknowledgments
- This work was supported by the Canada Excellence Research Chairs program (MJF). The Dr. Donald Rix BC Leadership Chair (MJF) is supported by Leading Edge Endowment Funds from the Province of British Columbia, LifeLabs and Genome BC. AnnEx is supported through the Canadian Consortium on Neurodegeneration in Aging. We also appreciate the Pacific Parkinson’s Research Institute, the Marg Meikle Professorship and the Mottershead Family gift (SA-C). We are very grateful for thoughtful discussions with Dr. Neil Cashman, Dr. Michael Coulthart and the Canadian CJD Surveillance System. We appreciate technical support from the Centre for Applied Neurogenetics, most especially Dan Evans and Omar Zabaneh for AnnEx development.
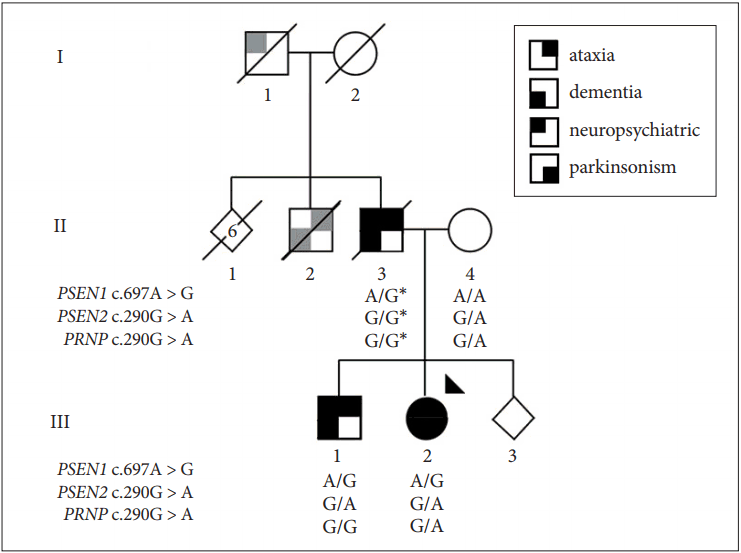
Figure 1.Pedigree of the family. The family’s presentation is consistent with dominant inheritance of a neurodegenerative disease. Symbols provide a segmented view of the most consistent clinical features reported [parkinsonism and ataxia, (lower or upper left quadrants), dementia and neuropsychiatric disturbance (lower and upper right quadrants)]. However, data on different symptom components may be incomplete and/or by history (gray quadrants). The proband analyzed by whole-exome sequencing is indicated by an arrowhead. Genotypes for the PSEN1, PSEN2 and PRNP mutations are reported. The (*) indicates inferred genotypes. PSEN1: presenilin 1, PSEN2: presenilin 2, PRNP: prion protein.
REFERENCES
- 1. Cruts M, Theuns J, Van Broeckhoven C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat 2012;33:1340–1344.ArticlePubMedPMC
- 2. Ryan NS, Rossor MN. Correlating familial Alzheimer’s disease gene mutations with clinical phenotype. Biomark Med 2010;4:99–112.ArticlePubMedPMC
- 3. Zheng L, Longfei J, Jing Y, Xinqing Z, Haiqing S, Haiyan L, et al. PRNP mutations in a series of apparently sporadic neurodegenerative dementias in China. Am J Med Genet B Neuropsychiatr Genet 2008;147B:938–944.ArticlePubMed
- 4. Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–315.ArticlePubMedPMCPDF
- 5. Ryan NS, Nicholas JM, Weston PSJ, Liang Y, Lashley T, Guerreiro R, et al. Clinical phenotype and genetic associations in autosomal dominant familial Alzheimer’s disease: a case series. Lancet Neurol 2016;15:1326–1335.ArticlePubMed
- 6. Mendez MF, McMurtray A. Frontotemporal dementia-like phenotypes associated with presenilin-1 mutations. Am J Alzheimers Dis Other Demen 2006;21:281–286.ArticlePubMedPMC
- 7. Houlden H, Crook R, Dolan RJ, McLaughlin J, Revesz T, Hardy J. A novel presenilin mutation (M233V) causing very early onset Alzheimer’s disease with Lewy bodies. Neurosci Lett 2001;313:93–95.ArticlePubMed
- 8. Guerreiro RJ, Baquero M, Blesa R, Boada M, Brás JM, Bullido MJ, et al. Genetic screening of Alzheimer’s disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol Aging 2010;31:725–731.ArticlePubMed
- 9. Wallon D, Rousseau S, Rovelet-Lecrux A, Quillard-Muraine M, Guyant-Maréchal L, Martinaud O, et al. The French series of autosomal dominant early onset Alzheimer’s disease cases: mutation spectrum and cerebrospinal fluid biomarkers. J Alzheimers Dis 2012;30:847–856.ArticlePubMed
- 10. Portet F, Dauvilliers Y, Campion D, Raux G, Hauw JJ, Lyon-Caen O, et al. Very early onset AD with a de novo mutation in the presenilin 1 gene (Met 233 Leu). Neurology 2003;61:1136–1137.ArticlePubMed
Citations
Citations to this article as recorded by

- Presenilin Gene Mutation-associated Psychosis
Mark A. Colijn, Zahinoor Ismail
Alzheimer Disease & Associated Disorders.2024; 38(1): 101. CrossRef - A heterozygous de novo PSEN1 mutation in a patient with early-onset parkinsonism
Yueting Chen, Peng Liu, Fei Xie, Bo Wang, Zhiru Lin, Wei Luo
Neurological Sciences.2022; 43(2): 1405. CrossRef - Nomenclature of Genetic Movement Disorders: Recommendations of the International Parkinson and Movement Disorder Society Task Force – An Update
Lara M. Lange, Paulina Gonzalez‐Latapi, Rajasumi Rajalingam, Marina A.J. Tijssen, Darius Ebrahimi‐Fakhari, Carolin Gabbert, Christos Ganos, Rhia Ghosh, Kishore R. Kumar, Anthony E. Lang, Malco Rossi, Sterre van der Veen, Bart van de Warrenburg, Tom Warner
Movement Disorders.2022; 37(5): 905. CrossRef - Progressive cognitive impairment and familial spastic paraparesis due to PRESENILIN 1 mutation: anatomoclinical characterization
Miren Altuna, Rosa Larumbe, María Victoria Zelaya, Sira Moreno, Virginia García-Solaesa, Maite Mendioroz, María Antonia Ramos, María Elena Erro
Journal of Neurology.2022; 269(9): 4853. CrossRef - Genetics, Functions, and Clinical Impact of Presenilin-1 (PSEN1) Gene
Jaya Bagaria, Eva Bagyinszky, Seong Soo A. An
International Journal of Molecular Sciences.2022; 23(18): 10970. CrossRef - Scoring Algorithm‐Based Genomic Testing in Dystonia: A Prospective Validation Study
Michael Zech, Robert Jech, Sylvia Boesch, Matej Škorvánek, Ján Necpál, Jana Švantnerová, Matias Wagner, Ariane Sadr‐Nabavi, Felix Distelmaier, Martin Krenn, Tereza Serranová, Irena Rektorová, Petra Havránková, Alexandra Mosejová, Iva Příhodová, Jana Šarlá
Movement Disorders.2021; 36(8): 1959. CrossRef - PET/MRI Delivers Multimodal Brain Signature in Alzheimer’s Disease with De Novo PSEN1 Mutation
Gayane Aghakhanyan, Dorothee Saur, Michael Rullmann, Christopher M. Weise, Matthias L. Schroeter, Ken Marek, Rami Abou Jamra, Solveig Tiepolt, Maria Strauss, Cordula Scherlach, Karl-Titus Hoffmann, Osama Sabri, Joseph Classen, Henryk Barthel
Current Alzheimer Research.2021; 18(2): 178. CrossRef - Spinocerebellar Ataxia-Like Presentation of the M233V PSEN1 Mutation
Yury Seliverstov, Ilya Kanivets, Sergey Illarioshkin
The Cerebellum.2020; 19(5): 744. CrossRef - NetCore: a network propagation approach using node coreness
Gal Barel, Ralf Herwig
Nucleic Acids Research.2020; 48(17): e98. CrossRef - Diagnostic Approach of Early-Onset Dementia with Negative Family History: Implications from Two Cases of Early-Onset Alzheimer’s Disease with De Novo PSEN1 Mutation
Jia Liu, Qianqian Wang, Donglai Jing, Ran Gao, Jing Zhang, Chunlei Cui, Hongwen Qiao, Zhigang Liang, Chaodong Wang, Pedro Rosa-Neto, Liyong Wu, Jianping Jia, Serge Gauthier
Journal of Alzheimer's Disease.2019; 68(2): 551. CrossRef - A Clinical Case of Patient Carrying Rare Pathological PSEN1 Gene Mutation (L424V) Demonstrates the Phenotypic Heterogenity of Early Onset Familial AD
Kaloyan R. Stoychev, Maya Stoimenova-Popova, Petranka Chumpalova, Lilia Ilieva, Mohamed Swamad, Zornitsa Kamburova-Martinova
Frontiers in Psychiatry.2019;[Epub] CrossRef - Deficiency in the transcription factor NRF2 worsens inflammatory parameters in a mouse model with combined tauopathy and amyloidopathy
Ana I. Rojo, Marta Pajares, Angel J. García-Yagüe, Izaskun Buendia, Fred Van Leuven, Masayuki Yamamoto, Manuela G. López, Antonio Cuadrado
Redox Biology.2018; 18: 173. CrossRef
 E-submission
E-submission

 PubReader
PubReader ePub Link
ePub Link Cite
Cite
