E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 11(1); 2018 > Article
-
Letter to the editor
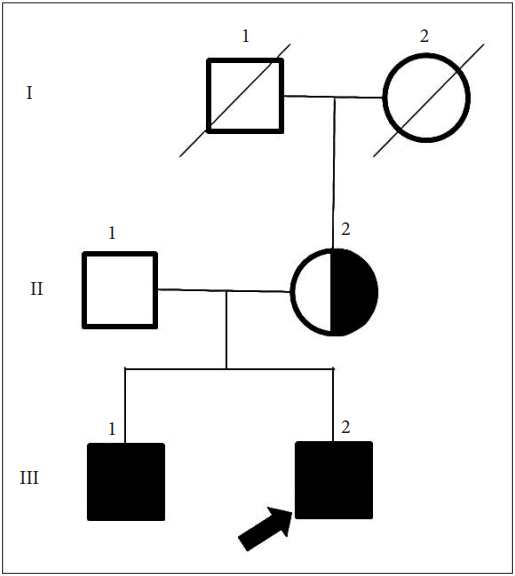
Myotonia Congenita Can Be Mistaken as Paroxysmal Kinesigenic Dyskinesia - Aryun Kim1, Mihee Jang1, Han-Joon Kim1, Yoon Kim1, Dae-Seong Kim2, Jin-Hong Shin2, Beomseok Jeon1
-
Journal of Movement Disorders 2018;11(1):49-51.
DOI: https://doi.org/10.14802/jmd.17056
Published online: January 23, 2018
1Department of Neurology, Seoul National University Hospital, Seoul National University College of Medicine, Seoul, Korea
2Department of Neurology, Pusan National University Yangsan Hospital, Yangsan, Korea
- Corresponding author: Beomseok Jeon, MD, PhD, Department of Neurology, Seoul National University Hospital, Seoul National University College of Medicine, 101 Daehak-ro, Jongno-gu, Seoul 03080, Korea / Tel: +82-2-2072-2876 / Fax: +82-2-3672-7553 / E-mail: brain@snu.ac.kr
• Received: September 7, 2017 • Revised: October 19, 2017 • Accepted: October 24, 2017
Copyright © 2018 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
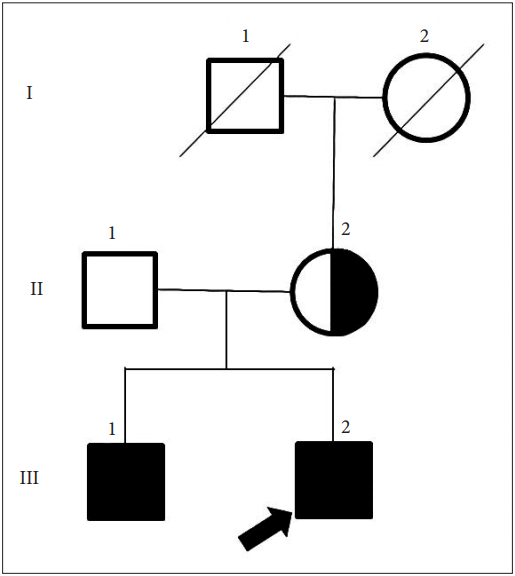
Figure & Data
References
Citations
Citations to this article as recorded by 
- Genetic updates on paroxysmal dyskinesias
James Y. Liao, Philippe A. Salles, Umar A. Shuaib, Hubert H. Fernandez
Journal of Neural Transmission.2021; 128(4): 447. CrossRef - A Japanese family with primary familial brain calcification presenting with paroxysmal kinesigenic dyskinesia - A comprehensive mutational analysis-
Akihiko Mitsutake, Takashi Matsukawa, Kristine Joyce L. Porto, Tatsuya Sato, Junko Katsumata, Tomonari Seki, Risa Maekawa, Takuto Hideyama, Masaki Tanaka, Hiroyuki Ishiura, Tatsushi Toda, Shoji Tsuji, Yasushi Shiio
Journal of the Neurological Sciences.2020; 418: 117091. CrossRef - Paroxysmal movement disorders – practical update on diagnosis and management
Claudio M. De Gusmao, Laura Silveira-Moriyama
Expert Review of Neurotherapeutics.2019; 19(9): 807. CrossRef - The study of exercise tests in paroxysmal kinesigenic dyskinesia
Hai-Yan Zhou, Fei-Xia Zhan, Wo-Tu Tian, Chao Zhang, Yan Wang, Ze-Yu Zhu, Xiao-Li Liu, Yang-Qi Xu, Xing-Hua Luan, Xiao-Jun Huang, Sheng-Di Chen, Li Cao
Clinical Neurophysiology.2018; 129(11): 2435. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite