Heterogeneous Patterns of Striatal Dopamine Loss in Patients with Young- versus Old-Onset Parkinson’s Disease: Impact on Clinical Features
Article information

Abstract
Objective
Ample evidence has suggested that age at onset of Parkinson’s disease (PD) is associated with heterogeneous clinical features in individuals. We hypothesized that this may be attributed to different patterns of nigrostriatal dopamine loss.
Methods
A total of 205 consecutive patients with de novo PD who underwent 18F-FP-CIT PET scans (mean follow-up duration, 6.31 years) were divided into three tertile groups according to their age at onset of parkinsonian motor symptoms. Striatal dopamine transporter (DAT) availability was compared between the old- (n = 73) and young-onset (n = 66) groups. In addition, the risk of developing freezing of gait (FOG) and longitudinal requirements for dopaminergic medications were examined.
Results
The old-onset PD group (mean age at onset, 72.66 years) exhibited more severe parkinsonian motor signs than the young-onset group (52.58 years), despite comparable DAT availability in the posterior putamen; moreover, the old-onset group exhibited more severely decreased DAT availability in the caudate than the young-onset group. A Cox regression model revealed that the old-onset PD group had a higher risk for developing FOG than the young-onset group [hazard ratio 2.523, 95% confidence interval (1.239–5.140)]. The old-onset group required higher doses of dopaminergic medications for symptom control than the young-onset group over time.
Conclusion
The present study demonstrated that the old-onset PD group exhibited more severe dopamine loss in the caudate and were more likely to develop gait freezing, suggesting that age at onset may be one of the major determinants of the pattern of striatal dopamine depletion and progression of gait disturbance in PD.
There is mounting evidence that Parkinson’s disease (PD) is a heterogeneous disorder rather than a single disease entity [1]. The identification of PD subtypes has received one of the highest priority recommendations in clinical research for PD because it may help to understand underlying disease mechanisms, predict disease course, and facilitate the design of more efficient personalized therapeutic strategies [2]. Consequently, several studies have attempted to delineate PD subtypes [3,4]; ample evidence has suggested that age at onset is a major determinant of clinical heterogeneity in patients with PD [4-7]. Although the age cut-offs used to define “young-onset PD” and “old-onset PD” vary among studies, the most consistent findings have been slower disease progression, better response to levodopa, more frequent motor complications, and less frequent cognitive impairment in patients with young-onset PD versus those with old-onset PD [5].
However, the exact mechanism underlying the age-at-onset-dependent differences in PD remains largely unknown. Given that different clinical subtypes of PD are associated with heterogeneous patterns of pathological involvement [8,9], there may be differences in the patterns of degeneration of dopaminergic neurons between patients with young- and old-onset PD. To date, few reports have demonstrated that age at onset is related to different patterns of striatal dopamine depletion in PD [10-13]; however, these studies were limited by small sample sizes [10], the confounding effects of PD medications [11,12], and less detailed segmentation of the striatum [10-13]. Therefore, in the present study, we compared the availability of striatal dopamine transporter (DAT) between patients with young- and old-onset de novo PD, with more detailed striatal segmentation. Additionally, we tested whether the age at onset of PD was associated with the rate of disease progression. To do this, we examined long-term motor outcomes in relation to the risk of developing freezing of gait (FOG) and in relation to the longitudinal requirement of dopaminergic medications over time according to the age at onset of PD.
MATERIALS & METHODS
Subjects
The medical records of 252 consecutive patients with de novo PD who visited the movement disorders outpatient clinic from April 2009 to June 2013 were reviewed. Of these, 47 patients who were lost to follow-up within 3 years were excluded. PD was diagnosed according to the clinical diagnostic criteria of the United Kingdom PD Society Brain Bank, and all patients exhibited decreased striatal DAT availability on 18F-N-(3-fluoropropyl)-2β-carbon ethoxy-3β-(4-iodophenyl) nortropane (18F-FP-CIT) positron emission tomography (PET) at the baseline evaluation. Parkinsonian motor symptoms were assessed using the Unified Parkinson’s Disease Rating Scale Part III (UPDRS-III). The Korean version of the Mini-Mental State Examination (K-MMSE) was used to assess general cognition. The doses of PD medications were calculated as levodopa-equivalent doses (LEDs) [14]. The patients were then classified according to the age at onset of parkinsonian symptoms. Given that age cut-offs to classify patients as either young-onset PD or old-onset PD have not yet been established [5], a total of 205 patients with de novo PD were subdivided into tertile groups according to their age at onset of their parkinsonian symptoms. Sixty-six patients were assigned to the young-onset PD group [the lowest tertile group (age at onset < 58 years)], and 73 were assigned to the old-onset PD group [the highest tertile group (age at onset > 66 years)]. We additionally recruited 20 age- and sex-matched healthy controls who underwent 18F-FP-CIT PET scans. The healthy controls were divided into the young control group (n = 10; mean age, 53.20 ± 2.82 years; female, 50.0%) and the old control group (n = 10; mean age, 71.40 ± 4.38 years; female 50.0%) and were compared with the respective age-matched PD groups. This study was approved by our Institutional Review Board (IRB no. 4-2014-0637). Given the retrospective nature of the present study, requirements for informed consent were waived.
Quantitative analyses of 18F-FP-CIT PET images
18F-FP-CIT PET was performed using a Discovery 600 (GE Healthcare, Milwaukee, WI, USA) device, which acquires images with a three-dimensional resolution of 2.3 mm full-width at half-maximum. After a 6-hour fast, subjects were intravenously injected with 5 mCi (185 MBq) of 18F-FP-CIT. Ninety minutes after the injection, PET images were acquired for 20 minutes in the three-dimensional mode, and spiral CT images were obtained for attenuation correction with 120 kVp and a current of 200 mA.
We performed quantitative analyses of 18F-FP-CIT PET data as previously described [15]. We performed image processing using SPM8 (Wellcome Department of Imaging Neuroscience, Institute of Neurology, UCL, London, United Kingdom) and MATLAB 2013a (MathWorks, Natick, MA, USA) for Windows. Volumes of interest (VOI) were defined based on the template in standard space. Reconstructed PET images were then spatially normalized to the Montreal Neurology Institute template space using a standard 18F-FP-CIT PET template, which was generated from T1-weighted magnetic resonance (T1 MR) and 18F-FP-CIT PET images of 13 healthy controls. Twelve VOI of the striatal subregions and one occipital VOI were drawn on a coregistered spatially normalized single T1 MR and 18F-FP-CIT PET template image using MRIcro version 1.37 (Chris Rorden, Columbia, SC, USA). The striatal subregions consisted of the anterior/posterior caudate, anterior/posterior putamen, ventral putamen, and ventral striatum. The boundaries of the striatal subregions were defined as described in our previous work [16]. These VOI were adjusted using a minor translation with the editing software ANIQUE [17]. DAT availability was calculated according to the nondisplaceable binding potential as follows: [mean standardized uptake value (SUV) of the striatal subregion VOI – mean SUV of the occipital VOI] / (mean SUV of the occipital VOI). We also calculated the intersubregional ratio for the anterior posterior gradient (i.e., caudate to posterior putamen).
Assessment of the development of FOG during the follow-up period
FOG is defined as an unintentional and temporary phenomenon in which the feet fail to progress forward despite the intention to walk [18], and its appearance has been associated with disease progression [19]. Various FOG subtypes have been identified: start hesitation; freezing on turning; freezing in restricted areas; destination freezing; and open space hesitation. Patients were asked about the development of FOG at every visit. Gait was also inspected (“medication on” status) at the outpatient clinic, where patients were specifically asked about the characteristic subjective sensation of the feet becoming “glued to the floor” in either the on or off state. The time from parkinsonian symptoms onset to the development of FOG was assessed using Kaplan-Meier estimates, and a log-rank test was used to compare the Kaplan-Meier curves between the PD groups. A Cox regression model was used to estimate the hazard ratio (HR) and corresponding 95% confidence interval (CI) while adjusting for sex, DAT availability (either in the posterior putamen or caudate), LED at the development of FOG or at last visit to the outpatient clinic, and K-MMSE scores. A log-minus-log plot and time-dependent covariate analysis revealed that the assumption of proportionality was reasonable.
Assessment of longitudinal changes in the dose of dopaminergic medications over time
In this study, patients with PD visited the outpatient clinic at three-month intervals for at least 3 years; the doses of PD medications were adjusted for effective symptom management according to patients’ response. A linear mixed model was used to compare the rate of longitudinal changes in LEDs between the two PD groups. Four fixed effects were included in the model: three were between-subject effects (PD group according to age at PD onset, sex, and baseline UPDRS-III scores), and one was a within-subject effect (time). Time was treated as a categorical variable with a 6-month interval (0 to 36) because most of the increases in LED occurred within the first 6 months, and then the doses of PD medications were adjusted every 3 months thereafter. The effects of the PD group on longitudinal changes in LED were tested using a time × PD group interaction term.
Statistical analyses
The baseline demographic characteristics of subjects in the PD groups were compared using Student’s t-tests and Pearson’s χ2 tests for continuous and categorical variables, respectively. To compare DAT availability in each striatal subregion, Student’s t-test with Bonferroni correction for multiple comparisons was used. The effects of age at PD onset on the development of FOG were assessed using a log-rank test and the Cox regression model as described above. A linear mixed model was used to compare the rate of longitudinal changes in LED between the PD groups. Statistical analyses were performed using SPSS version 23.0 (IBM Corp., Armonk, NY, USA); statistical significance was set at p < 0.05 (two-tailed).
RESULTS
Baseline clinical characteristics and striatal DAT availability in patients with PD
The patients in the old-onset PD group [mean (± SD) age at onset of PD, 72.66 ± 4.71 years; mean age at PD diagnosis, 73.80 ± 4.94 years] were older than those in the young-onset PD group (51.15 ± 5.06 years, p < 0.001; age at PD diagnosis, 52.58 ± 5.00 years, p < 0.001). Patients in the old-onset PD group had higher UPDRS-III scores (24.26 ± 10.01) than those in the young-onset PD group (20.79 ± 9.68; p = 0.040). The old-onset PD group had lower K-MMSE scores (26.77 ± 1.78) than the young-onset PD group (27.58 ± 2.02; p = 0.013). There were no significant differences in sex or disease duration between the groups (Table 1).

Demographic characteristics of patients with PD
DAT availability in each striatal subregion in the PD groups is also presented in Table 1. The old-onset PD group exhibited more severely decreased DAT availability in the caudate than the young-onset PD group (anterior caudate, 1.91 ± 0.58 vs. 2.50 ± 0.70, p < 0.001; posterior caudate, 1.19 ± 0.40 vs. 1.55 ± 0.51, p < 0.001), while DAT availability in the other striatal subregions (anterior putamen, posterior putamen, ventral putamen, and ventral striatum) did not differ between the groups. The old-onset PD group showed a lower ratio of caudate to posterior putamen (1.46 ± 0.41) than the young-onset PD group (1.85 ± 0.47; p < 0.001). Additionally, there were no significant differences in DAT availability of all striatal subregions between the old and young control groups (Supplementary Table 1 in the online-only Data Supplement). The old control group also had a lower ratio of caudate to posterior putamen (0.82 ± 0.03) than the young control group (0.87 ± 0.07; p = 0.036), but the decrease in the ratio in the control groups (-5.7%) was much smaller than the decrease in the PD groups (-21.1%).
The effect of age at onset of PD on the development of FOG
During the follow-up period, FOG developed in 14 of 66 patients in the young-onset PD group and in 24 of 73 in the old-onset PD group. FOG occurrence over time in the PD groups is shown in Figure 1. Kaplan-Meier analysis revealed that the old-onset PD group had a higher risk for developing FOG than the young-onset group (PLog-rank = 0.007). The Cox regression model revealed that patients in the old-onset PD group had a higher risk for developing FOG than those in the young-onset PD group [HR 2.523, 95% CI (1.239–5.140), p = 0.011, while adjusting for DAT availability in the posterior putamen; HR 2.511, 95% CI (1.175–5.366), p = 0.017, while adjusting for DAT availability in the caudate] (Table 2). Additionally, when the Cox regression model was applied to the entire 205 patients with PD, with age at PD onset as a continuous variable, the age at onset significantly affected the development of FOG [HR 1.041, 95% CI (1.010–1.074), p = 0.010, while adjusting for DAT availability in the posterior putamen; HR 1.042, 95% CI (1.008–1.078), p = 0.016, while adjusting for DAT availability in the caudate], while the striatal DAT availability did not [posterior putamen, HR 1.033, 95% CI (0.514–2.074), p = 0.928; caudate, HR 1.033, 95% CI (0.568–1.878), p = 0.916; Supplementary Table 2 in the online-only Data Supplement]. Thus, if a patient with PD exhibited parkinsonian symptoms one year later, the patient had an approximately 4% higher risk for developing FOG.

Kaplan-Meier estimates curves of the development of FOG after onset of parkinsonian symptoms in patients with youngonset PD (n = 66) and old-onset PD (n = 73). The old-onset group demonstrated a higher risk for developing FOG than the young-onset group (PLog-rank = 0.007). The crosses in the plots indicate censored data. FOG: freezing of gait, PD: Parkinson’s disease.
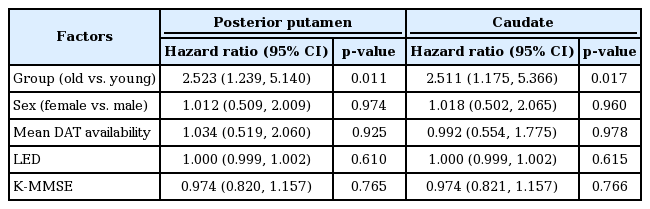
Cox regression analysis for the development of freezing of gait according to the age at onset of parkinsonism
The effect of age at onset of PD on the rate of longitudinal changes in LED
There was a significant time × PD group interaction in the mixed model (p < 0.001), indicating that the pattern of longitudinal changes in LED differed between the young- and old-onset PD groups. Throughout the follow-up period, the old-onset PD group required higher doses of PD medications than the young-onset PD group for effective control of parkinsonian symptoms (Table 3, Figure 2).

Longitudinal changes in levodopa-equivalent dose
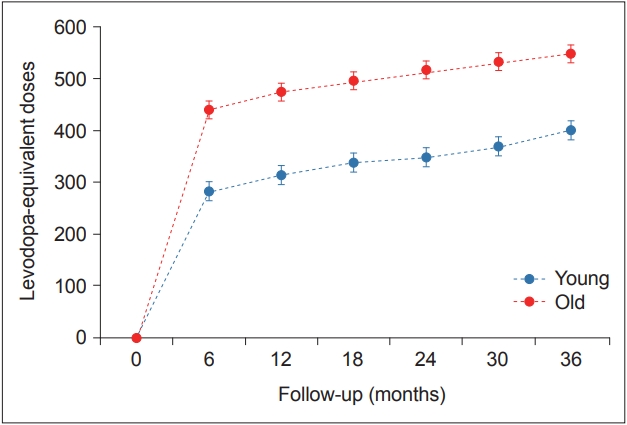
Longitudinal increases in levodopa-equivalent doses. The young-onset group received lower doses of dopaminergic medications for symptom control than the old-onset group. There was a significant interaction between groups and time in the mixed model (group × time: p < 0.001).
DISCUSSION
In the present study, we investigated the effects of age at PD onset on the patterns of striatal dopamine depletion and long-term motor outcomes in relation to the risk for developing FOG and longitudinal changes in LED over time. The major findings are summarized as follows: 1) Patients in the old-onset PD group had higher UPDRS-III scores than those in the young-onset PD group, despite comparable DAT availability in the posterior putamen; 2) The old-onset PD group exhibited more severely decreased DAT availability in the caudate than the young-onset PD group, while DAT availability in the other striatal subregions did not differ between the groups; 3) Patients in the oldonset PD group had a higher risk for developing FOG than those in the young-onset PD group; and 4) The old-onset PD group required higher doses of dopaminergic medications than the young-onset PD group throughout the follow-up period. These findings suggest that the onset of parkinsonian symptoms at older ages is associated with relatively diffuse striatal dopamine depletion and poorer long-term motor outcomes.
Selective dopaminergic denervation in the posterior putamen is the principal pathological feature of PD [20], although the exact pathogenic mechanisms underlying this regional selectivity remain unclear. Aside from the posterior putamen, other striatal subregions are also gradually affected in PD to various degrees with uncertain clinical relevance. Postmortem studies have reported that patients with PD accompanied by disabling tremor exhibited greater dopamine neuronal loss in the retrorubral area (A8) [9] but less in the substantia nigra pars compacta (A9) [8] compared to those without tremor as a dominant parkinsonian symptom. Several PET studies have demonstrated that the extent of involvement of extra-putaminal subregions varies among patients with PD [21,22] and have suggested a link between striatal dopamine loss pattern and clinical heterogeneity [12,23,24]. Given that striatal dopamine deficiency represents the primary dysregulation of neurotransmitter action in PD [25] and that the striatum can be subdivided according to topographically and functionally organized connections [26], patterns of striatal dopamine depletion may reflect the clinical phenotypes of PD.
In this regard, a few pathological and PET/single-photon emission computed tomography studies have explored the relationship between age at onset, clinical heterogeneity, and patterns of nigrostriatal dopamine deficit in patients with PD. A postmortem study involving 12 young- and 22 old-onset cases of PD reported greater nigral cell loss in the young-onset PD group, without differences in basic Lewy body pathology [27]. Two molecular imaging studies reported that patients with young-onset PD endured more damage to the nigrostriatal dopamine system at symptom manifestation than those with old-onset PD, suggesting more efficient compensatory mechanisms in younger individuals [10,11], while other studies found no such association between age at onset and the severity of nigrostriatal damage [28-30]. Recently, Liu et al. [12] reported that the ratio of caudate/putamen DAT binding was lower in the old-onset PD group than in the young-onset PD group, although absolute DAT binding in the caudate and putamen did not differ between the groups. Pagano et al. [13] also demonstrated more widespread dopaminergic dysfunction in elderly patients by analyzing data from the Parkinson’s Progression Marker Initiative (PPMI) database. In the present study, we classified patients with PD based on the distribution of age at onset in our database, and the cut-off ages to assign the patients to the young-onset PD group (i.e., 58 years) were much higher than in previous studies (i.e., most studies set the cut-off at 50 years of age). Nevertheless, our results are consistent with two recent studies [12,13] that reported more sparing of other striatal subregions than of the putamen in young-onset PD. This striatal dopamine loss pattern could be associated with some benign clinical features in younger patients, such as lower UPDRS-III scores and a slower rate of longitudinal increases in LED. In fact, some evidence has suggested that residual dopamine in the spared associative/limbic striatum would compensate for dopaminergic deficits in the sensorimotor striatum (i.e., posterior putamen) through sprouting of the remaining dopaminergic fibers or increased dopamine release and diffusion [31]. In addition, DAT availability in the caudate might be linked to cognitive function in the PD groups [32]. Although it remains elusive how age at onset affects the pattern of striatal dopamine loss in PD, some aging-dependent biological variables, such as neuronal progenitor proliferation, trophic environment, inflammation, and proteasome/lysosome function [33], would be possible explanations.
In this study, patients with old-onset PD had a higher risk for developing FOG than the younger-onset patients. Given that FOG mostly occurs in the advanced stages of PD and that its appearance is associated with disease progression [19], our findings support the concept that older age at onset is related to rapid disease progression [4]. In fact, the risk factors for developing FOG have yet to be established; in particular, the effect of age at onset on the occurrence of FOG remains controversial [34-37]. Moreover, the underlying pathophysiology of FOG remains poorly understood, although some evidence has suggested that the dopaminergic component is a critical element for the development of FOG [38]. Further studies are needed to determine the associations among age at onset, patterns of striatal dopamine loss, and the development of FOG in patients with PD. In addition, the old-onset PD group exhibited a more rapid rate of increases in LED than the young-onset PD group. Because the dose of dopaminergic medications is indirectly associated with parkinsonian disability [39], this result again suggests that age at onset is a key determinant of the rate of disease progression.
Some limitations of this study should be addressed. First, the patients were subdivided into tertile groups according to the distribution of age at onset, which may be somewhat arbitrary and may act as a bias. A consensus for the cut-off of ages to classify patients into young- versus old-onset groups is needed. Second, FP-CIT PET may not be an ideal surrogate marker for nigrostriatal dopaminergic degeneration [40]. Third, the occurrence of FOG was not assessed using objective gait measures or validated questionnaires, which may have led to an inaccurate estimation of FOG. Fourth, there is a possibility that atypical parkinsonism cases (e.g., progressive supranuclear palsy with predominant parkinsonism) might be misdiagnosed as idiopathic PD, although we made a thorough diagnosis based on the appropriate criteria and evaluation. Finally, due to the lack of follow-up data on Hoehn and Yahr stage or UPDRS-III scores, we assessed the clinical progression based on the longitudinal changes in LED, which might not accurately reflect the motor status of patients with PD.
In conclusion, the present study demonstrated that subjects in the old-onset PD group exhibited more severe dopamine loss in the caudate and were more likely to develop gait freezing. These findings suggest that age at onset may be a major determinant of the pattern of dopamine depletion and progression of gait disturbance in patients with PD.
Supplementary Materials
The online-only Data Supplement is available with this article at https://doi.org/10.14802/jmd.18064.
Supplementary Table 1.
Comparison of striatal DAT availability between young and old healthy controls
Supplementary Table 2
Cox regression analysis for the development of freezing of gait
Notes
Conflicts of Interest
The authors have no financial conflicts of interest.
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (grant number: NRF-2018R1D1A1B07048959).
The authors are grateful to Jungsu S. Oh and Jae Seung Kim (Department of Nuclear Medicine, Asan Medical Center, University of Ulsan College of Medicine) for the quantitative analyses of the 18F-FP-CIT PET images.