 E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 13(2); 2020 > Article
-
Case Report
Dopa-Responsive Dystonia: A Male Patient Inherited a Novel GCH1 Deletion from an Asymptomatic Mother -
Wendi Wang1,2
 , Baozhong Xin1, Heng Wang1,3,4
, Baozhong Xin1, Heng Wang1,3,4 -
Journal of Movement Disorders 2020;13(2):150-153.
DOI: https://doi.org/10.14802/jmd.19069
Published online: March 18, 2020
1DDC Clinic Center for Special Needs Children, Middlefield, OH, USA
2Department of Pediatrics, Vanderbilt University School of Medicine, Nashville, TN, USA
3University Hospitals Rainbow Babies & Children’s Hospital, Cleveland, OH, USA
4Department of Cardiovascular & Metabolic Sciences, Cleveland Clinic, Cleveland, OH, USA
- Corresponding author: Heng Wang, MD, PhD DDC Clinic Center for Special Needs Children, 14567 Madison Road, Middlefield, OH 44062, USA / Tel: +1-440-632-1668 / Fax: +1-440-632-1697 / E-mail: wang@ddcclinic.org
Copyright © 2020 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Dopa-responsive dystonia (DRD) is a complex genetic disorder with either autosomal dominant or autosomal recessive inheritance, with autosomal dominant being more frequent. Autosomal dominant DRD is known to be caused by mutations in the GCH1 gene, with incomplete penetrance frequently reported, particularly in males. Here, we report a male patient with DRD caused by exon 1 deletion in the GCH1 gene inherited from the asymptomatic mother. The patient had an atypical presentation, notably with no dystonia, and underwent extensive workup for a myriad of neuromuscular disorders before a low-dose L-dopa trial and confirmatory genetic testing were performed. Our experience with this family highlights an atypical presentation of DRD and prompts us to consider the genetic complexity of DRD.
- This patient was first presented to DDC Clinic in 2003 at the age of 5 years, with ongoing muscle weakness, urinary retention and incontinence, and constipation. His medical history prior to presentation was extensive. He was born full term after an uneventful pregnancy. He sat at 9 months but was unable to hold himself erect while sitting and would often slouch to the side or forward. He did not crawl and started walking at approximately 17 months. His parents described him as a “toe-walker” with a long history of frequent falls. At age 2, he developed chronic constipation requiring the use of laxatives to aid with bowel movements. He was also seen by a urologist for urinary retention and incontinence. At age 3, he continued to have difficulty using his legs and presented to pediatric neurologists for evaluation and workup. Although there were significant concerns about his gross motor development, his parents did not feel he had difficulty with cognitive or fine motor skills.
- MRI of his brain and spinal cord at age 3 suggested a possible tethered spinal cord, and he underwent neurosurgery to release the tethered cord shortly afterwards. However, even with this procedure and extensive post-procedure physical therapy, his motor functioning remained unimproved, and he continued to have urinary symptoms along with constipation. He progressively worsened and lost endurance in standing and walking, and it would take some time for him to regain endurance after an acute illness. He also started to exhibit drooling and facial and upper body weakness and was relying on a wheelchair for his daily activities. A second MRI of his brain and spine was performed at age 5, which revealed a normal myelination pattern but detected syrinxes at C5–C7 and T4–T5. This finding, however, did not seem to explain his symptoms given his predominant lower extremity weakness. Family history was not significant, although the patient’s mother reported that she was also a toe-walker as a child.
- On exam, he was wheelchair-bound with decreased muscle tone and muscle weakness in all extremities, more prominent in the legs. His sensation was fully intact with normal deep tendon reflexes, and no dystonia was noted on examination. Routine hematological and biochemical assays, including creatine phosphokinase, were all within normal ranges. Karyotyping, chromosome microarray, and extensive genetic and metabolic workup, including spinal muscular atrophy-related genetic testing, histologic, and biochemical assays of the muscle biopsy, were all unremarkable.
- Although he did not have the typical clinical profile of DRD, he was found to have notable diurnal fluctuation with alleviation of symptoms in the morning and aggravation of symptoms toward the evening even when he was 3 years old. He would be able to walk in the morning without his braces but slowly lost his strength as the day progressed. Near the end of the day, he would need assistance for sitting erect and eating his dinner. After repeatedly extensive workup without a definitive diagnosis, a levodopa trial to rule out DRD was proposed to the family. A low dose of levodopa (50 mg twice a day) was initiated at the age of 9 with a dramatic response. He was able to stand up from the wheelchair shortly after the trial started. He gradually regained motor skills and has remained symptom-free since. At the age of 21, he is able to participate in many sport activities, such as basketball, swimming and even cross country running, with a minimal increase in the dose of levodopa (a total of 125 mg a day) from its initiation.
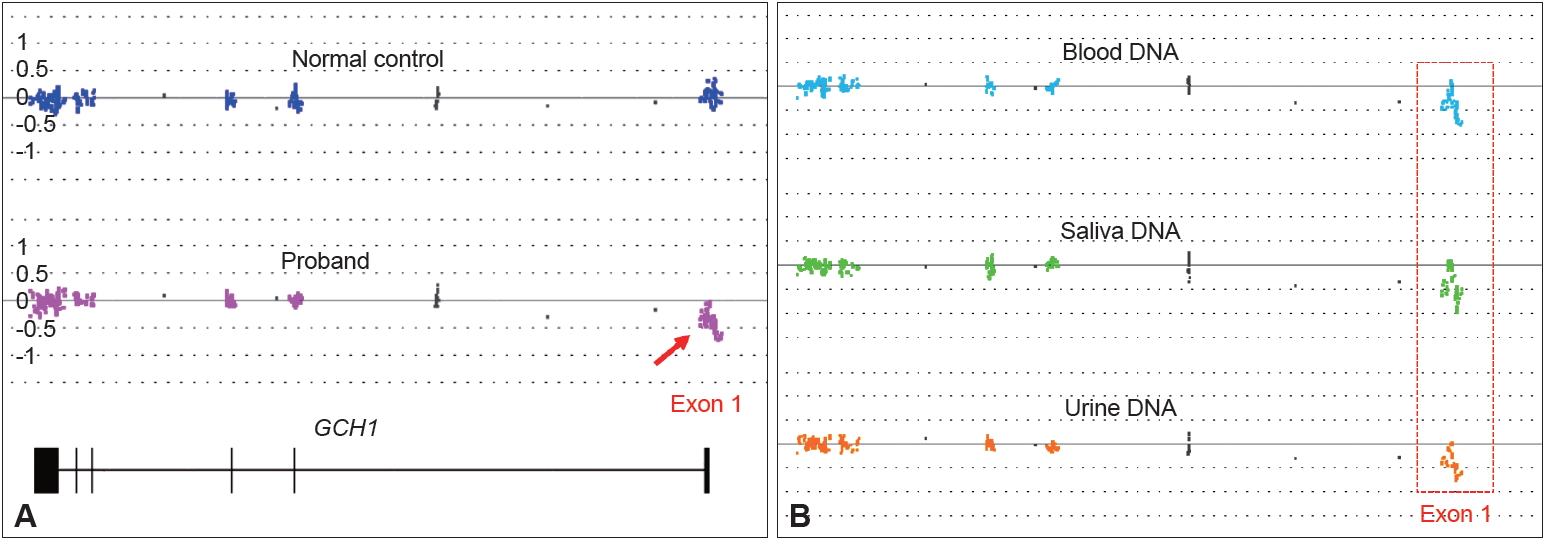
- Genetic analysis of the proband focusing specifically on the GCH1 gene was performed retroactively. Genomic DNA isolated from peripheral blood was analyzed through whole-gene sequencing and array-based exon-level copy number variation analysis. No pathogenic variant was detected through DNA sequencing. Microarray analysis identified a large single copy deletion encompassing exon 1 (Figure 1A). Loss of the entire exon 1 is predicted to lead to a null allele of GCH1. Heterozygous deletions in the GCH1 gene ranging from single exons to the whole gene have been reported previously in patients with DRD. This variant has not been observed in the control genomic variant databases. Based on the American College of Medical Genetics and Genomics (ACMG) standards and guidelines for the interpretation of sequence variants, it is classified as a pathogenic variant. Further DNA analysis revealed that this deletion was inherited from the mother of the proband (Figure 1B). The father and the unaffected sibling were negative for the deletion.
CASE REPORT
- DRD presents with a wide clinical spectrum but is typically associated with childhood-onset dystonia, gait disturbance, marked diurnal fluctuations with aggravation of symptoms in the evening and improvement with sleep or rest, and a dramatic and sustained response to low doses of L-dopa without motor fluctuations or dyskinesias as the hallmark of the disease [4]. Symptom onset usually occurs between ages 1 and 12 years, with a mean age of 6 years. Dystonia usually starts in the lower extremities and gradually becomes generalized. Individuals generally have normal cognitive function with unaffected cerebellar, sensory, and autonomic function. The condition can often result in misdiagnosis for cerebral palsy, but these patients are typically unresponsive to a variety of modalities used for treatment of cerebral palsy [5].
- Early diagnosis and treatment of DRD is extremely rewarding, as the dramatic response to levodopa typically seen in most patients results in significant improvements not only in motor disturbances but also in psychiatric and even sleep symptoms. It is often a completely life-changing event. However, the diagnosis of DRD is challenging, particularly for patients with atypical presentations. The patient we presented here had no significant dystonia but showed other atypical presentations, such as decreased muscle tone, autonomic dysfunction with urinary retention, incontinence and constipation, which were seldom reported in previous cases. The case was further complicated by other possible neurological disorders, such as tethered spinal cord, which introduced additional challenges. A low-dose L-dopa trial here was key for our patient’s diagnosis and might be considered in other similar cases. It is also interesting to note that urinary symptoms resolved soon after the initial L-dopa treatment was started in the patient, indirectly confirming these symptoms as being part of the disease. However, his constipation was not improved after levodopa treatment until he was in college, which allowed us to assume that constipation might not be a part of the DRD phenotype.
- Although DRD caused by GCH1 mutations is known to be inherited in an autosomal dominant fashion, the literature shows that the manifestation of this mutation can be quite variable. Previous observations have suggested female gender predominance among DRD patients, with a female to male ratio of 4.3:1; additionally, the extent of penetrance of GCH1 mutations is 2.3 times higher in females than in males [6], and one study even suggests almost complete penetrance in females [3]. Additional literature has suggested that females exhibit classic DRD symptoms more frequently and earlier in life than males, while males appear more likely to have a milder phenotype or later disease onset [2]. The aforementioned patient is unique, as he is an affected male with two unaffected parents. To our surprise, the asymptomatic mother was positive for the exon 1 deletion, which was passed onto the proband and caused the disease. Since the patient’s maternal grandfather was deceased with no DNA available and the maternal grandmother was negative for the deletion, we were not able to determine if the mutation was inherited or de novo in the asymptomatic mother. However, we found the same heterozygous deletion in her genomic DNAs collected from her saliva and urine samples, indicating a broad tissue distribution and thereby reducing the possibility of mosaicism in the mother.
- We present this unique case in which a female carrier is phenotypically asymptomatic for a disease that has been documented with almost 100% penetrance in females. To our knowledge, there have not been any reports to date describing an instance of an affected male inheriting a pathogenic variant from an asymptomatic mother, suggesting that the genetics underlying DRD remain complex and worthy of further study.
DISCUSSION
- We thank the patient and his family for their patience and support. We appreciate the many physicians who provided outstanding care to the patient.
- The study was supported in part by The Elisabeth Severance Prentiss Foundation, The Reinberger Foundation, The Leonard Krieger Fund of the Cleveland Foundation (L2009-0078), and The William Bingham Foundation.
Acknowledgments
-
Author Contributions
Conceptualization: Heng Wang. Data curation: all authors. Formal analysis: all authors. Funding acquisition: Baozhong Xin and Heng Wang. Investigation: all authors. Methodology: Baozhong Xin and Heng Wang. Project administration: Heng Wang. Resources: Heng Wang. Supervision: Heng Wang. Writing— original draft: all authors. Writing—review & editing: Baozhong Xin and Heng Wang.
Notes
- 1. Furukawa Y. GTP cyclohydrolase 1-deficient dopa-responsive dystonia. 2002 Feb 21 [Updated 2019 Jan 24]. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al., editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; c1993-2019. [accessed on 2019 Aug 10]. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1508/.
- 2. Trender-Gerhard I, Sweeney MG, Schwingenschuh P, Mir P, Edwards MJ, Gerhard A, et al. Autosomal-dominant GTPCH1-deficient DRD: clinical characteristics and long-term outcome of 34 patients. J Neurol Neurosurg Psychiatry 2009;80:839–845.ArticlePubMed
- 3. Steinberger D, Weber Y, Korinthenberg R, Deuschl G, Benecke R, Martinius J, et al. High penetrance and pronounced variation in expressivity of GCH1 mutations in five families with dopa-responsive dystonia. Ann Neurol 1998;43:634–639.ArticlePubMed
- 4. Lee WW, Jeon BS. Clinical spectrum of dopa-responsive dystonia and related disorders. Curr Neurol Neurosci Rep 2014;14:461.ArticlePubMedPMCPDF
- 5. Kulshreshtha D, Maurya PK, Singh AK, Thacker AK. Dopa-responsive dystonia in a child misdiagnosed as cerebral palsy. J Pediatr Neurosci 2017;12:172–173.ArticlePubMedPMC
- 6. Furukawa Y, Lang AE, Trugman JM, Bird TD, Hunter A, Sadeh M, et al. Gender-related penetrance and de novo GTP-cyclohydrolase I gene mutations in dopa-responsive dystonia. Neurology 1998;50:1015–1020.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
- Detection of Single-Nucleotide and Copy Number Defects Underlying Hyperphenylalaninemia by Next-Generation Sequencing
Elisabetta Anna Tendi, Giovanna Morello, Maria Guarnaccia, Valentina La Cognata, Salvatore Petralia, Maria Anna Messina, Concetta Meli, Agata Fiumara, Martino Ruggieri, Sebastiano Cavallaro
Biomedicines.2023; 11(7): 1899. CrossRef - Study on Mechanism of Cumulative Directional Blasting of Brittle Karst Limestone in the Guizhou Province
Jie Hu, Yiping Zhang, Chengcheng Fang, Yusong Miao, Xin Zhao, Dengguo Liu, José António Fonseca de Oliveira Correia
Advances in Materials Science and Engineering.2023; 2023: 1. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite
