1Department of Neurology, Seoul National University Hospital, Seoul, Korea
2Department of Neurology, Kyung Hee University Hospital, Seoul, Korea
Corresponding author: Beomseok Jeon, MD, PhD Department of Neurology, Seoul National University Hospital, Seoul National University College of Medicine, 101 Daehak-ro, Jongno-gu, Seoul 03080, Korea / Tel: +82-2-2072-2876 / Fax: +82-2-3672-7553 / E-mail: brain@snu.ac.kr
• Received: October 28, 2019 • Revised: December 9, 2019 • Accepted: January 28, 2020
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Niemann-Pick disease type C (NPC) is an autosomal recessive neurodegenerative disorder caused by mutation of either NPC1 (> 95% of cases) or NPC2 [1,2]. Mutations in these genes lead to massive accumulation of cholesterol and glycosphingolipids within late endosomes and lysosomes [1]. NPC is characterized by heterogeneity in age at onset, which is related to variance in clinical presentation and progression [2]. Patients with NPC are classified into 5 subgroups on the basis of age at onset: pre/perinatal (< 2 months), early-infantile (2 months to < 2 years), late-infantile (2 years to < 6 years), juvenile (6 years to 15 years), and adolescent/adult (> 15 years) [1,2].
Adult-onset NPC shows a wide spectrum of neurological symptoms such as vertical supranuclear gaze palsy (VSGP), ataxia, dystonia, cognitive decline and psychosis, and less commonly visceral involvement [3]. In clinical practice, adult-onset NPC is easily misdiagnosed or neglected due to rarity and clinical heterogeneity of this condition [4]. In particular, late adult-onset NPC is very rare, and thus its clinical characteristics remain unclear [2]. Early diagnosis of NPC is important because treatment with miglustat has been shown to stabilize disease progression [2]. Herein, we present a patient with late adult-onset NPC who had onset of symptoms at 57 years of age.
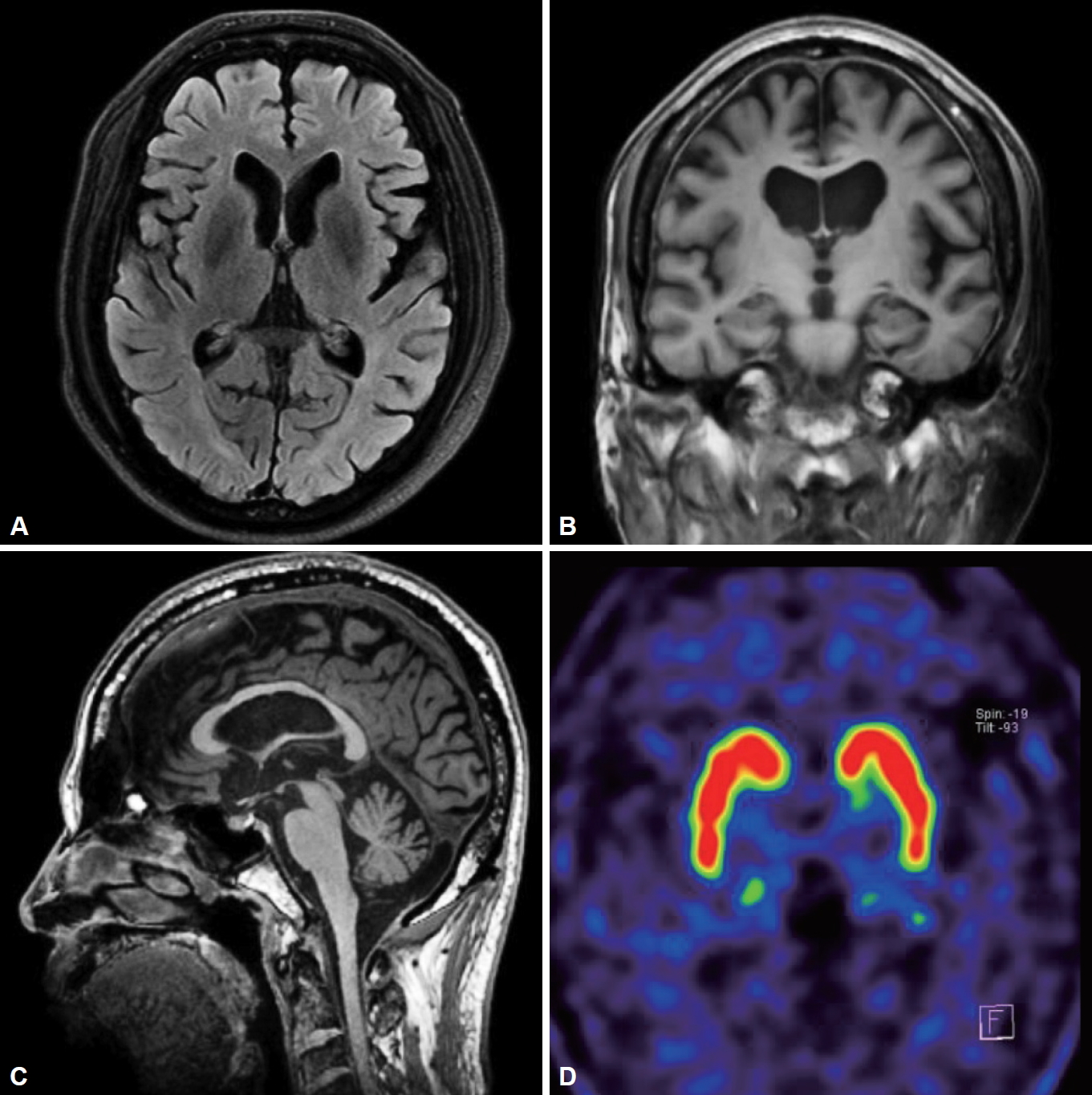
A 63-year-old man visited Seoul National University Hospital due to his slowly progressive gait disturbance and difficulties with speech and swallowing that began 6 years ago. He also presented with a one-year history of cognitive decline. The patient had no history of previous neurological and psychiatric disorders. There was no relevant family history. On neurologic examination, he showed vertical gaze limitations that were more pronounced in downgaze (Supplementary Video 1 in the online-only Data Supplement). There were no definite features suggestive of parkinsonism. The finger-to-nose test and heel-to-shin test showed dysmetria. He showed a mild wide-based gait with postural instability in turning (Supplementary Video 1 in the online-only Data Supplement). Tandem gait was impaired. The scores on the Mini-Mental State Examination, Montreal Cognitive Assessment, and Frontal Assessment Battery were 25, 24, and 13, respectively. Abdominal examination did not reveal any visceromegaly, which was confirmed by abdominal ultrasound. His clinical symptoms suggested the possibility of NPC or progressive supranuclear palsy with predominant cerebellar ataxia (PSP-C). The NPC suspicion index score [4] was 100, indicating its high probability. Brain magnetic resonance imaging showed diffuse brain atrophy (Figure 1A–C). There were no abnormalities in 18F-FP-CIT positron emission tomography (Figure 1D). Ataxia gene panel testing showed that NPC was positive for 2 pathogenic variants (p.A518T and p.A1059G) of the NPC1 gene [5]. The other genetic tests were all negative (Supplementary Table 1 in the online-only Data Supplement).
Our patient represents a unique case of NPC developing clinical symptoms at 57 years of age. Clinical features of this patient strongly suggest NPC. The genetic testing was also compatible with the recently proposed diagnostic criteria for NPC in which the diagnosis can be confirmed by the identification of two known or likely pathogenic alleles [1]. Although PSP-C should have been considered in this case, the patient showed preserved dopamine transporter uptake. Furthermore, despite 6 years of disease duration, his symptoms were relatively mild. These findings suggest that the patient is less likely to have PSP-C. In Korea, Lee et al. [6]. first described two siblings diagnosed with adolescent/adult-onset NPC in their mid-20s. We believe that this is the second case report of adolescent/adult-onset NPC and the first case report of late adult-onset NPC in Korea.
A large international prospective registry including 163 NPC patients showed that adolescent/adult-onset accounted for 27% of the patients [7]. In that study, the mean age (standard deviation) of symptom onset in adolescent/adult-onset group was 29.7 (10.0) years, implying that individuals with adolescent/adult-onset NPC mainly develop initial symptoms during the third or fourth decade. However, NPC can present in the sixth decade, although it is extremely rare [2]. To the best of our knowledge, the onset of symptoms in our patient is only the second case reported with symptom onset at 59 years of age [3].
In adult-onset NPC, diagnostic delay is common and often 5 years or more, as seen in our case. This delay may be minimized when VSGP is identified. Although VSGP can be observed in other neurological disorders, it is one of the earliest signs in NPC. VSGP is present in approximately 70–80% of the patients across all age at onset categories [2], which suggests that clinicians should consider NPC in the differential diagnosis of VSGP, regardless of age at onset. However, despite the diagnostic utility of VSGP, it may not be present when the patients are examined early in the disease course [1]. Thus, the absence of VSGP cannot rule out the diagnosis of NPC, and eye movements need to be checked regularly in patients with suspected NPC, but without VSGP.
Video 1. The first part (1–23 seconds) shows reduced vertical gaze that was more pronounced in downgaze. The second part (24–36 seconds) shows a mild wide-based gait with postural instability in turning, but his arm swing and walking speed were preserved. The last part (37 onwards) shows impaired tandem gait.
Notes
Conflicts of Interest
The authors have no financial conflicts of interest.
Author Contributions
Conceptualization: Ryul Kim and Beomseok Jeon. Data curation: Ryul Kim and Dallah Yoo. Supervision: Beomseok Jeon. Visualization: Ryul Kim. Writing— original draft: Ryul Kim. Writing—review & editing: Dallah Yoo, Sangmin Park, Jung Hwan Shin, Ji-Hyun Choi, Han-Joon Kim, and Beomseok Jeon.
Acknowledgments
None.
Figure 1.
Neuroimaging findings. Brain magnetic resonance imaging reveals diffuse cortical atrophy on an axial fluid-attenuated inversion recovery image (A) and a coronal T1-weighted image (B) and midbrain and cerebellar atrophy on a sagittal T1-weighted image (C). 18F-FP-CIT positron emission tomography shows normal presynaptic uptake (D).
REFERENCES
1. Geberhiwot T, Moro A, Dardis A, Ramaswami U, Sirrs S, Marfa MP, et al. Consensus clinical management guidelines for Niemann-Pick disease type C. Orphanet J Rare Dis 2018;13:50.ArticlePubMedPMCPDF
2. Kumar N, Rizek P, Mohammad Y, Jog M. Pearls & Oy-sters: Niemann-Pick disease type C in a 65-year-old patient. Neurology 2016;87:e79–e81.ArticlePubMedPMC
3. Sévin M, Lesca G, Baumann N, Millat G, Lyon-Caen O, Vanier MT, et al. The adult form of Niemann-Pick disease type C. Brain 2007;130(Pt 1):120–133.ArticlePubMedPDF
4. Wijburg FA, Sedel F, Pineda M, Hendriksz CJ, Fahey M, Walterfang M, et al. Development of a suspicion index to aid diagnosis of Niemann-Pick disease type C. Neurology 2012;78:1560–1567.ArticlePubMed
5. Imrie J, Heptinstall L, Knight S, Strong K. Observational cohort study of the natural history of Niemann-Pick disease type C in the UK: a 5-year update from the UK clinical database. BMC Neurol 2015;15:257.ArticlePubMedPMC
6. Lee SY, Lee HJ, Kim SH, Jeong YJ, Jin HK, Bae JS, et al. Two siblings with adolescent/adult onset Niemann-Pick disease type C in Korea. J Korean Med Sci 2016;31:1168–1172.ArticlePubMedPMC
7. Patterson MC, Mengel E, Wijburg FA, Muller A, Schwierin B, Drevon H, et al. Disease and patient characteristics in NP-C patients: findings from an international disease registry. Orphanet J Rare Dis 2013;8:12.ArticlePubMedPMC
Figure & Data
References
Citations
Citations to this article as recorded by
Lysosomal storage disorders identified in adult population from India: Experience of a tertiary genetic centre and review of literature Jayesh Sheth, Aadhira Nair, Riddhi Bhavsar, Koumudi Godbole, Chaitanya Datar, Sheela Nampoothiri, Inusha Panigrahi, Heli Shah, Shruti Bajaj, Naresh Tayade, Naveen Bhardwaj, Harsh Sheth JIMD Reports.2024; 65(2): 85. CrossRef
Genetic and phenotypic variability in adult patients with Niemann Pick type C from Serbia: single-center experience Nikola Kresojević, Valerija Dobričić, Milica Ječmenica Lukić, Aleksandra Tomić, Igor Petrović, Nataša Dragašević, Ivana Perović, Ana Marjanović, Marija Branković, Milena Janković, Ivana Novaković, Marina Svetel, Vladimir S. Kostić Journal of Neurology.2022; 269(6): 3167. CrossRef
Two Patients with Niemann Pick Disease Type C Diagnosed in the Seventh Decade of Life Melanie Wu, Rita Ceponiene, Ece Bayram, Irene Litvan Movement Disorders Clinical Practice.2020; 7(8): 961. CrossRef
 E-submission
E-submission
 , Dallah Yoo2
, Dallah Yoo2
 PubReader
PubReader ePub Link
ePub Link Cite
Cite