 E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 13(2); 2020 > Article
-
Review Article
Long-Term Outcomes of Genetic Parkinson’s Disease -
Jan O. Aasly1,2

-
Journal of Movement Disorders 2020;13(2):81-96.
DOI: https://doi.org/10.14802/jmd.19080
Published online: May 29, 2020
1Department of Neurology, St. Olav’s Hospital, Trondheim, Norway
2Department of Neuroscience, Norwegian University of Science and Technology, Trondheim, Norway
- Corresponding author: Jan O. Aasly, MD, PhD Department of Neurology, St Olavs Hospital, Edvard Griegs gate 8, 7006 Trondheim, Norway / Tel: +47 7257 5071 / Fax: +47 7257 5774 / E-mail: Jan.Aasly@ntnu.no
• Received: October 30, 2019 • Revised: February 7, 2020 • Accepted: March 23, 2020
Copyright © 2020 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Parkinson’s disease (PD) is a progressive neurodegenerative disorder that affects 1–2% of people by the age of 70 years. Age is the most important risk factor, and most cases are sporadic without any known environmental or genetic causes. Since the late 1990s, mutations in the genes SNCA, PRKN, LRRK2, PINK1, DJ-1, VPS35, and GBA have been shown to be important risk factors for PD. In addition, common variants with small effect sizes are now recognized to modulate the risk for PD. Most studies in genetic PD have focused on finding new genes, but few have studied the long-term outcome of patients with the specific genetic PD forms. Patients with known genetic PD have now been followed for more than 20 years, and we see that they may have distinct and different prognoses. New therapeutic possibilities are emerging based on the genetic cause underlying the disease. Future medication may be based on the pathophysiology individualized to the patient’s genetic background. The challenge is to find the biological consequences of different genetic variants. In this review, the clinical patterns and long-term prognoses of the most common genetic PD variants are presented.
- PubMed searches were performed using key words and strings, such as Parkinson’s disease, parkinsonism, monogenic, genetic, survival, mortality, follow-up, and long term, in combination with each gene or variants known to cause PD in a small percentage of cases. The historical nomenclature starting with PARK and followed by a number has been kept for relevant mutations.
- SNCA/PARK1
- The mutations causing PARK1 PD are located on chromosome 4 in SNCA, which codes for the protein α-synuclein. α-Synuclein is abundant in the brain, and smaller amounts are found in the heart, muscles, and other tissues. The function of α-synuclein is not well understood. It is suggested that it plays an important role in maintaining an adequate supply of synaptic vesicles in presynaptic terminals, exocytosis, the regulation of neurite outgrowth, and nerve cell adhesion.
- PARK1 was the first gene that could be related to familial PD. Families located in southern Italy demonstrated an autosomal-dominant trait of typical PD through many generations followed back several centuries. The locus was found in 1996, and one year later, the mutation was shown to be in the α-synuclein gene at chromosome 4 [18]. Although this is a rare cause of PD, it is of great importance for understanding the pathology of the disease. It was soon shown that LBs, the hallmark of PD, contain large amounts of α-synuclein [1,19,20]. The Contursi kindred could trace their ancestry back to a couple who had lived in the late 1600s. The members shared the same A53T mutation in SNCA, and they were all probably related through a common ancestor. Later, additional missense SCNA mutations were shown to cause PD, such as E46K, H50Q, G51D and A53E. In some other families, duplications and triplications of the α-synuclein gene have been found to be rare causes of hereditary PD or PD-like syndromes [21-26]. In homozygous duplication cases, the phenotypes are characterized by early-onset and rapidly progressive parkinsonism followed by dementia, which has been seen to be more aggressive in cases of SNCA triplication [27].
- Most patients with SNCA-related PD present with early and asymmetric onset, initial good response to dopaminergic therapy and early motor complications. A 20-year follow-up was performed among members of the Contursi family [28]. A relevant variability was observed in the combination and severity of motor symptoms and nonmotor features. Age at onset (AAO) varied from 26 to 48 years (mean, 32 years), with longer disease duration than previously reported. Cognitive deficits were present in all patients, ranging from frank dementia in those with longer disease duration to moderate cognitive impairment or slight isolated executive dysfunction in those with shorter disease duration. Depression and anxiety were detected in most patients, and behavioral disorders and dysautonomia were present in half of them. Olfaction was impaired in all tested patients. Hyposmia, constipation and REM sleep behavior disorder are all markers for this type of PD and are often referred to as synuclein markers. No mitochondrial defect was detected in extensive biochemical evaluations [29].
- The “Iowa kindred,” a large Iowan family with autosomal-dominant PD, has been followed clinically since the 1920s at the Mayo Clinic. In 2003, the genetic cause was determined to be a 1.7 Mb triplication of the α-synuclein genomic locus. Affected individuals presented with an early-onset, severe parkinsonism-dementia syndrome [30,31]. Microsatellite variability within the genomic interval was identical to that previously described for a Swedish-American family with an α-synuclein triplication. Subsequent genealogical investigation suggested that both kindreds were ancestrally probably related to the Lister family complex [26].
- Three family members of a Canadian kindred carrying a heterozygous A53E mutation in SNCA had moderate to severe bradykinesia with rigidity, postural instability, freezing of gait, dyskinesia, and myoclonus, including spontaneous myoclonus. The AAO varied from 25 and 37 to 58 years, with postural myoclonus, focal reflex myoclonus after hand stimulation, and generalized reflex myoclonus. All three individuals developed dementia after a few years [32]. Although cognitive dysfunction was not described for Finnish A53E carriers, it has been observed in some individuals with the A53T α-synuclein mutation [33]. There was no cognitive decline among the Finnish patients through the relatively short period of follow-up, which was for 5 years only [33]. They did not share the same allele, showing that these were two distinct families.
- The effect of subthalamic nucleus deep brain stimulation (STN-DBS) in SNCA/PARK1 patients is probably comparable to what is reported in idiopathic PD patients, with no major adverse events, satisfactory improvement of motor function and a reduction of pharmacological treatment, although the total number of operated patients is low [34].
- Although the penetration of disease may vary, SNCA/PARK1 patients usually have severe parkinsonism with rapid progression and the development of dementia. The patients with triplication have on average an earlier onset and a more progressive disease than the patients with duplication and those carrying a single mutation. On average, patients with amplifications of or point mutations in SNCA have a severe PD phenotype. At autopsy, these patients show abundant LB pathology.
- PRKN/PARK2-related PD
- The PRKN/PARK2 gene encodes the E3-ubiquitin ligase parkin. E3-ubiquitin ligases are involved in the proteasome pathway, which permits the degradation of damaged target proteins by ubiquitin adjunction, and parkin is also essential for maintaining mitochondrial homeostasis [35]. PRKN is the most common cause of autosomal-recessive PD and accounts for almost 50% of typical early-onset parkinsonism. Mutations are highly diverse, including missense mutations and nonsense mutations, frameshifts, and rearrangements with exon deletions or multiplications, but all of them lead to protein loss of function or the absence of protein by nonsense mRNA decay. Recently, the number of PRKN disease-causing mutations reached almost 140. This number will probably rise when all new cost-effective, technologically advanced sequencing techniques are implemented and more patients from different ethnic groups are tested. In a recent review of monogenic PD patients from multiple centers around the world, patients with mutations in PRKN made up approximately 80–85% of autosomal-recessive cases. In comparison, patients with PRKN mutations were half as common as patients with either LRRK2 or GBA mutations [17].
- The mean AAO of PRKN/PARK2-related PD is approximately 30 years, ranging from childhood to over 50 years in some rare cases. PRKN mutations are responsible for 77% of cases of juvenile PD, with an AAO before 21 years. Patients present with a typical PD phenotype with the clinical triad and a good response to levodopa. There are, however, differences between patients with PRKN/PARK2-related PD and those with sporadic PD. PRKN patients more often have lower-limb dystonia and more symmetrical symptoms at onset [3]. Patients with PRKN mutations may present with hyperreflexia and early motor fluctuations.
- Furthermore, there are no phenotypical differences between patients with PRKN missense mutations and those with disruptive mutations. Although mitophagy is regulated by PINK1 and parkin proteins, it is quite paradoxical that no PD patients have a better prognosis than those with PD caused by PRKN or PINK1 mutations. The disease progression is very slow, with a sustained response to levodopa. Abrupt and severe fluctuations are common, which makes medical treatment quite challenging. Only a very few patients have cognitive decline; the patients with PRKN mutations usually remain cognitively unaffected and do not show typical PD dementia [36,37]. Additional features such as psychiatric manifestations are rare, and olfaction function is well preserved [38]. Dysautonomia and other atypical features are also rare. Heterozygous PRKN mutations may be a risk factor for PD, although with an age of onset at much higher age than in homozygous carriers [39]. Heterozygous PRKN and PINK1 mutation carriers may show mild PD, being less progressive and responsive to L-dopa administration [40].
- Long-term survival of patients with PRKN-related PD is usually favorable. Although they have a low age at disease onset, most of them have a long disease duration without dementia [36,41]. The good prognosis of patients with PRKN and PINK1 mutations and parkinsonism has led to the creation of the term “nigropathy,” indicating less deterioration of cognitive function [37]. Patients with PRKN mutations usually respond well to levodopa therapy, but many patients experience rapid fluctuations with sudden changes in motor function. The freezing of gait, postural deformity, and motor fluctuations are common late features with severe dyskinesias, and sudden on-off episodes are common complications [42].
- Four family members suffering from young-onset PRKN-related PD, located in the Middle East, were followed for 40 years [41]. The patients all had beneficial responses to levodopa, slow progression of the disease, and diurnal fluctuations. They shared similar clinical features to other patients with PRKN-related PD, including AAO, symptoms at disease onset, beneficial response to levodopa, and the occurrence of nonmotor symptoms. However, some features were regarded as unique to that kindred: all members had hypersensitivity to levodopa, responding dramatically to very low doses of levodopa, and they were treated with strikingly low doses of levodopa. The age of onset ranged from 18 to 39 years, which is a typical variation also seen in other families with PRKN-related PD. Lower-limb dystonia, often observed among patients with PRKN-related PD, was not observed in that family.
- An Italian group performed a multicenter, case-control study to investigate the prevalence and severity of impulse-control disorders (ICDs) in a cohort of patients with PRKN-related PD. They compared their group of patients with PD patients without PRKN mutations, matched for demographic and clinical features. They collected 22 PRKN-related PD patients with a mean follow-up of 22 years [43]. Although the occurrence of ICDs did not correlate with the parkin genotype, parkin mutations influenced both the onset and the severity of specific behavioral disturbances in the impulsive-compulsive spectrum. In particular, ICDs were overall more severe in PRKN mutation carriers, who also disclosed a higher frequency and severity of compulsive shopping, binge eating, and punding. Regarding hypersexuality, patients with parkin-related PD reported a similar frequency but a more severe clinical expression. Moreover, ICDs tended to occur earlier in patients with the PRKN mutation, sometimes predating PD onset. Their results also suggested an association between the PRKN genotype, smoking status, and ICD severity [43].
- Depression and anxiety may be common in early-onset PD. A multicenter study of PRKN-related PD patients sought to determine the relation between depression, as evaluated by two instruments, the Patient Health Questionnaire and the Beck Depression Inventory II, and genotype in a family-based sample of probands with early-onset PD, EOPD, and their relatives. They did not find any positive association of depression with genotype among EOPD probands, although they had a relatively high prevalence of depression, indicating depressed mood for at least some days to a week over the last two weeks [44]. DBS has become an important part of the therapeutic armamentarium in complicated PD cases. There are many short reports each describing the favorable effect of DBS on small groups of PRKN-related PD patients, and there are some case reports with similar conclusions [45-47]. Rizzone et al. [48] recently published an excellent review of the outcome of DBS in patients with a genetic background of PD. The PRKN-related PD patients generally had a favorable outcome with DBS. As in other types of PD, the best results were observed in patients with a good response to levodopa, a younger age and no or few axial, non-levodopa-responsive motor symptoms. The sudden changes in motor func tions may still create extra challenges to PRKN-related PD patient treatment after DBS surgery [36].
- The major difference between sporadic PD and PRKN-related PD is seen upon autopsy. Patients with PRKN-related PD present neuronal loss predominantly in the ventral substantia nigra, and so far, only a few of them have shown LBs. The selectivity of the lesions could explain the lack of cognitive decline. There is a disparity in the severity of nigral loss between patients with sparse LBs and those with no LBs in patients with PRKNrelated PD, supporting the notion that abnormal α-synuclein deposition is not an integral component of the pathology of PRKN-related PD [36,49]. In a relatively large series of patients with PRKN-related PD and a review of the literature, together with autopsy reports of 13 patients with PRKN-related PD, only 3 out of 13 had any LBs. Neuronal loss in the substantia nigra was observed in all 13 patients [42].
- The disease progression in patients with PRKN-related PD is very slow, with a sustained response to levodopa. Abrupt and severe fluctuations are common, which makes medical treatment quite challenging. Patients with PRKN-related PD usually remain cognitively unaffected and do not show typical PD dementia [36,37]. Additional features such as psychiatric manifestations are rare, and olfaction function is well preserved [38]. Dysautonomia and other atypical features are also rare. These individuals are probably more prone to develop ICDs. The selectivity of the lesions could explain the lack of cognitive decline. Furthermore, there are no phenotypical differences between patients with PRKN missense mutations and those with disruptive mutations.
- PINK1/PARK6-related PD
- PINK1 encodes PTEN-induced putative kinase 1. PINK1 is the second most frequent gene involved in autosomal-recessive PD, and PINK1-related PD has a typical phenotype and early onset. PINK1 is almost as frequent as PRKN in North Africa. The PINK1-related PD phenotype is similar to that of the PRKN-related PD phenotype, with a slightly later AAO, in the early thirties, a good response to levodopa and rare cognitive decline. However, some differences can be noted, with less spasticity, pyramidal signs or hyperreflexia than patients with PRKN mutations. To date, 60 mutations of different types (missense, nonsense, splicing, frameshift and deletions) have been reported. In contrast to patients with PRKN-related PD, patients with PINK1-related PD may show neuronal loss in the substantia nigra and the presence of LBs.
- Kasten et al. [50] performed an extensive review of 139 PINK1 mutation carriers from 85 families. In contrast to sporadic PD, the proportion of men in the group with PINK1-related PD was less than that of women, 42%. The PINK1-related cases were distributed all over the world, and the majority of patients were of Caucasian ethnicity, followed by mixed/other and Asian ethnicity [50]. A majority of patients were from Italy (20%), probably because Italian scientists were the first to describe the mutation [4]. There was a considerable proportion also from Iran (10%) and from Spain (8%).
- The median AAO of all PINK1 mutation carriers was 32 years, with the majority (62%) having an early, 22% a late, and 15% a juvenile PD AAO [50]. It was long debated whether heterozygous mutation carriers could be at risk for developing PD, although with a later onset [51,52]. These early observations have not been reproduced, and reports from Tunisia, where PINK1 mutations are very common, indicate that being a heterozygous mutation carrier does not increase the risk for PD [53].
- The long list of disease-generating mutations is listed in the paper by Kasten, including a complete listing of the frequencies of signs and symptoms in patients with PINK1-related PD [50]. Eighty-three percent of all patients carried a homozygous mutation, and 17% of these were compound heterozygous mutations. They had all kinds of mutations, including missense mutations, the most frequent type, in 47.6%, followed by structural variants in 19.1% and nonsense mutations in 14%. The most frequent mutation of all was the missense mutation c.1040T>C, resulting in an amino acid change of leucine to proline at position 347. As with most other PD-related genes, the disease-causing potential of PINK1 genes varies. Only one-fifth of the mutations are classified as definitely pathogenic; the rest are classified as probably or possible pathogenic.
- Dyskinesia was reported in 39% of the patients, dystonia in one-fifth, and motor fluctuations in one-third of the patients. Levodopa-induced dyskinesias were very common. Cognitive decline and psychotic symptoms were reported in a small fraction of all patients with PINK1-related PD. The majority of patients with PINK1-related PD show excellent or very good response to levodopa therapy.
- There are many reports on PINK1-related PD in the literature, but there are very few long-term follow-up studies. Five affected PINK1 homozygous and 14 heterozygous mutation carriers from two large Italian families were followed over a 12-year period [54]. Motor, nonmotor, cognitive, psychiatric, and behavioral profiles were systematically assessed. Four homozygotes and eight heterozygotes underwent magnetic resonance imaging. All homozygotes showed a mild progression of motor signs and a persistent excellent response to levodopa. All but one patient complained of nonmotor symptoms and sleep impairment. Three presented with ICDs and two with anxiety and apathy. All obtained abnormal scores on the Montreal Cognitive Assessment (MoCA) and tests sensitive to frontal lobe functions; one presented global cognitive impairment. Three heterozygotes showed motor signs and were diagnosed as possibly affected. They had nonmotor symptoms and cognitive impairment, and two of them showed mild bilateral temporal atrophy [54]. This long-term follow-up did not confirm severe hyposmia or impaired color discrimination, as suggested by earlier studies [55,56].
- These findings are somewhat in contrast to my own experiences with patients with PINK1-related PD. In my study of four patients with homozygous or compound heterozygous mutations, the AAO was between 28 and 44 years, with a mean age of 36 years. They have been repeatedly tested over a mean period of 15 years. Their phenotype has been quite identical to that of patients with classic PD. The presenting symptom was unilateral tremor with additional bradykinesia and rigidity. They have had good or excellent levodopa effects, and after some years, they have developed dyskinesias and fluctuations, comparable to what is regularly seen in individuals with classic PD. One of them had mild neuropsychiatric complaints, mainly anxiousness. They all tested normal on cognitive tests and remained cognitively unaffected many years after disease onset.
- Several studies have shown that patients with PINK1 mutations benefit from deep brain stimulation, either in the subthalamic nuclei or in the bilateral globus pallidus internus (GPi). In a study with 5 PKRN- and PINK1-related PD patients, the clinical response was similar to that observed in nonmutation carriers. These patients have slightly more advanced axial motor symptoms at a relatively early disease stage [46,57]. This confirms my own experiences with good and maintained effects of STN-DBS and GPi DBS in patients with PINK1-related PD.
- One of my patients with PINK1-related PD was autopsied at age 79 years. At the age of 76, her MoCA score was 30/30 after 32 years of illness duration. One year later, she was taken to the hospital after falls and infection. Her health gradually deteriorated, and she died in a nursing home two years later. At autopsy, there was typical neuronal loss in the substantia nigra with LBs in the brainstem and in the cerebral cortex. There were no amyloid deposits (to be published). This is in accordance with two other single cases of PINK1-related PD autopsies. The first case showed neuronal loss in the substantia nigra pars compacta, LBs and aberrant neurites in the reticular nuclei of the brainstem, substantia nigra pars compacta and Meynert nucleus, but the locus coeruleus and the amygdala were spared [58]. This was similar to what we observed in my patient. The second autopsy report was from an 85-year-old woman with more complex disease, testing positive for a homozygous PINK1 mutation [59]. Her brain pathology was, in principle, not different from that of the abovementioned cases.
- The PINK1-related PD phenotype is characterized by slow disease progression, and the response to levodopa is good. The sense of smell is less likely to be less affected than in sporadic PD [31]. Psychiatric comorbidity and gait disturbance are common in PINK1-related PD compared to PRKN-related PD. This PD variant showed substantia nigra pars compacta neuronal loss, LBs and aberrant neuritis in the brainstem, with amygdala sparing. Although mitophagy is regulated by PINK1 and parkin proteins, it is quite paradoxical that no PD patients have a better prognosis than those with PD caused by PRKN or PINK1 mutations.
- DJ-1/PARK7-related PD
- Since the first published report of mutations in the oncogene DJ-1, few patients with DJ-1 mutations have been reported, but it still remains the third most frequent autosomal-recessive early-onset PD-related gene after PRKN and PINK1 [50,60,61]. The gene is localized to chromosome 1p36 and comprises seven coding exons encoding a DJ-1 protein of 189 amino acids. The DJ protein is expressed in both cerebral and extracerebral tissues and plays a role as a cellular sensor of oxidative stress [62]. Twenty different disease-causing sequence variants have been reported in the DJ gene, and there are no ethnicity-specific differences [50]. The median AAO of all patients was 27 years (22–35 years), with the majority (83%) having an early AAO, 13% having a juvenile AAO, and 4% having a late AAO of PD [50]. To date, there has been a majority of male cases [50]. Studies from several parts of the world have demonstrated that this is a rare cause of PD [63-66]. Among patients with early-onset PD, the prevalence of DJ-1 related PD varies between 0.4% and 1% [63]. Patients share the same phenotype as those with PRKN- or PINK1-related PD, but compared to them, more nonmotor signs, including depression, cognitive decline, psychosis and anxiety, have been reported in patients with DJ-1-related PD [61].
- Most patients with DJ-1-related PD respond to levodopa therapy, while others may be quite therapy resistant [67]. In a study from India in patients with early-onset autosomal-recessive PD, PARK2 mutations were most common, accounting for approximately 77% of familial and approximately 20% of EOPD patients in general. DJ-1 mutations were rare, accounting for 1–2% of sporadic cases of EOPD in India [68].
- There have been only a very few reports on neuropathology in PARK7-related PD. They all indicate that this is an α-synucleinopathy associated with LBs [67]. Patients with DJ-1-related PD usually have more severe anosmia and peripheral synucleinopathy than patients with parkin- and PINK1-related PD [50,69]. To date, there have been few reports on DBS in DJ-1-related PD patients [48,70].
- DJ-1-related PD is a very rare, early-onset, recessive form of PD. Its phenotype is similar to that seen in PKRN- and PINK1-related PD. Most patients respond well to levodopa. One report indicates good outcome from DBS treatment [48].
- LRRK2/PARK8-related PD
- The PARK8 locus was described by Funayama et al. [71] in 2002 when analyzing a large Japanese multi-incident PD family. They localized the gene to chromosome 12p11.2-q13.1. Several other groups searched the same area, which led to the discovery of mutations in that locus in 2004 [6,7]. A large Norwegian family could be traced back to 1,580 (Figure 1).
- The case of another large multi-incident family had been published prior to the identification of the gene. These results showed that family members with identical genetic backgrounds may have typical PD genotypes with quite different pathoanatomical findings at autopsy [72]. Several other mutations were later discovered, as was the most common, G2019S [73,74]. The original Japanese family was later found to carry the I2020T mutation [75].
- The LRRK2 gene encodes a 286 kDa multidomain peptide whose actions include neurite outgrowth, cytoskeletal maintenance, vesicle trafficking, the regulation of autophagy, and immune functioning [76]. More than 100 LRRK2 gene variants have been reported, most of which are “rare orphan changes,” and their significance is unknown [77]. At least eight LRRK2 mutations are known to increase the risk of familial PD [78]. These mutations are in the gene’s 1) kinase, 2) Ras of complex protein (ROC)/ GTPase, and 3) C-terminal of Ras (COR) domains (Figure 2) [79]. A recent analysis concluded that G2019S, A419V, R1441C/G/H, N1437H, V1699C and I2020T are pathogenic mutations and that G2385R and R1628 are variants significantly associated with increased risk for PD, while the R1398H mutation was associated with a significantly decreased risk for PD [80-82]. The G2019S mutation, considered to be the most common PD-associated “pathogenic substitution” LRRK2 mutation [83], increases LRRK2 protein kinase (phosphorylation) activity. The frequency of this mutation is highest among Ashkenazi Jews and in North African Arab-Berber populations. Several studies have shown that 15% of PD patients with Ashkenazic Jewish heritage carry this mutation [84,85]. Estimates of the lifetime penetrance of this mutation have varied from 22% to 100% [73,86,87]. A study co-authored by the International LRRK2 Consortium found the risk of PD for G2019S mutation carriers to be 28% at age 59 years, 51% at 69 years, and 74% at 79 years [86]. The discrepancies between the different estimates of the penetrance of G2019S may be due in part to different populations being studied; the highest estimates were from multi-incident families, whereas lower penetrance was found in ethnically diverse PD populations not selected on the basis of familial history [88]. The G2385R and R1628P mutations are susceptibility (risk) variants, rather than penetrant variants, and are found in 3–4% of healthy individuals and 6–8% of PD patients in some Asian populations [81,89,90]. Recent studies have shown that the G2385R mutation reduces LRRK2 kinase activity [91,92], destabilizes LRRK2 and promotes its proteasomal degradation [92]. The N1437H, R1441C, R1441G, Y1699C, and I2020T mutations are less common mutations and are found in certain areas and ethnic groups [86].
- Independent of mutations, genome-wide association study approaches have also identified LRRK2 as a common genetic risk factor for sporadic PD [94]. Large-scale genotyping and gene sequencing of LRRK2 have identified risk factors associated with PD. One example is LRRK2 p.G2385R as a risk factor in Asian populations [95]. There is also evidence of a haplotype in LRRK2 that is inversely associated with PD and other Parkinsonplus syndromes. LRRK2 parkinsonism has pleomorphic pathology [96]. The precise mechanism by which variations around the LRRK2 gene region contribute to disease risk is not fully determined. However, given that the polymorphisms associated with sporadic PD are in the promoter region of LRRK2, a reasonable hypothesis is that these variants do not change protein structure or function but instead alter the expression levels of the gene. A number of studies support this hypothesis, demonstrating increased LRRK2 protein in the brain [97] and blood [79,98] and increased LRRK2 kinase activity in the brain [99] and urine [100,101] in patients with sporadic PD compared with controls.
- Several studies have shown that LRRK2 plays an important role in vesicular trafficking, impacting endosomal, lysosomal, and autophagosomal pathways [102]. Importantly, numerous other PD-related genes, including SNCA and VPS35, also converge on these closed loop control pathways, suggesting that these substrates are interacting in the disease pathophysiology. The G2019S mutation may play a role in α-synuclein and/or tau pathology in LRRK2-related PD because α-synuclein in LBs is extensively phosphorylated, and phosphorylated tau colocalizes with α-synuclein in some LBs. Phosphorylation is thought to increase tau neurotoxicity, but the influence of phosphorylation on α-synuclein toxicity is unclear. Although some studies have suggested that LRRK2 may phosphorylate α-synuclein and/or tau [103-105], the only presently accepted theory is that the substrates for LRRK2 kinase are LRRK2 protein itself and some Rab GTPases such as Rab10.105 The current view is that when expressed in the same cell, in disease, α-synuclein and LRRK2 likely interact in response to mitochondrial disruption, in autophagy pathways in the endolysosomal system, and in normal interactions with common protein partners such as 14-3-3 proteins that maintain the homeostasis of a number of systems [105]. Targeting LRRK2 as a therapeutic strategy is supported by recently published studies that have demonstrated that genetically knocking out LRRK2106 or chronic administration of an LRRK2 kinase inhibitor in rats107 has been shown to protect against dopamine neuron loss induced by viral-mediated α-synuclein overexpression. This is of vital importance when planning future therapies for retarding disease progression. Preclinical studies have confirmed that the inhibition of LRRK2 may be protective against neuron loss [108]. This implies that antisense therapy could be aimed at either LRRK2 or α-synuclein expression both in PD and in multiple system atrophy.
- Interestingly, the mean delay before treatment may be shorter in sporadic PD patients than in LRRK2 patients: 3 years vs. 4 years in LRRK2 patients. Studies in asymptomatic LRRK2 mutation carriers have shown that the premotor or preclinical period may last many years. Multi-incident families with autosomal LRRK2-related PD have, for the first time, made it possible to study the development of PD many years prior to phenoconversion. PET-scan studies with different amine isotopes in asymptomatic mutation carriers have shown that there are upregulations in dopamine, serotonin and choline receptors in the asymptomatic period [109-111]. Since only a few of these carriers convert to PD, it is still impossible to estimate the length of the mean premotor or prodromal phase in LRRK2-related PD.
- Most patients with LRRK2-related PD show clinical features indistinguishable from those with classic sporadic PD. In the longitudinal The Parkinson’s Progression Markers Initiative study, most patients with LRRK2-related PD had cardinal signs and symptoms that were not different from those with sporadic PD. Patients with LRRK2 G2019S mutations have slightly better smell performance than those with sporadic PD [112,113]. This may reflect a lower burden of LBs and a better prognosis in the majority of cases.
- To date, there have been only a few prospective studies on LRRK2-related PD. The largest study spanning 9 years was conducted in 3 centers where they followed patients of Jewish ethnicity with PD with LRRK2 G2019S mutations [114]. The groups were rather identical, and the LRRK2-related PD group had a lower increase in Unified Parkinson’s Disease Rating Scale (UPDRS) scores than the control group of patients with sporadic PD. The change in MoCA scores was lower in the LRRK2 group, but the difference did not reach statistical significance [114]. These results were in accordance with the results from a multicenter cross-sectional survey performed more than 10 years ago [86]. These were later confirmed in small studies from different ethnic groups [115,116]. One study also showed that carriers of the G2019S mutation converted to PD 10 years earlier in a population from the Middle East than in a population from Northern Europe [117]. Genetic studies have indicated that this difference in phenoconversion may be based on genetic modifiers and not on environmental factors [118]. There have been no studies on survival comparing patients with LRRK2-related PD to those with sporadic PD, and there are no systematic studies on progression among LRRK2 gene mutations other than G2019S. There is one study among carriers of LRRK2 variants. In a study among carriers of risk variants G2385R, R1628P, and S1647T and noncarriers, motor score progression was defined as the difference between UPDRS motor scores at baseline and at follow-up. A total of 184 patients, 122 risk variant carriers and 62 noncarriers, were evaluated and followed up for up to 6.5 years. The risk variant carriers experienced a greater rate of motor progression than noncarriers after 4 years from the date of diagnosis, suggesting that these risk variants may facilitate neurodegeneration with increasing disease duration [119].
- There have been a number of studies trying to find good biomarkers for PD progression by using preclinical patients with LRRK2-related PD and comparing these to patients with LRRK2-related PD, patients with sporadic PD and normal controls.
- Analytes examined in these studies included Aβ1-42, tau, α-synuclein, oxidative stress markers, autophagy-related proteins, pteridines, neurotransmitter metabolites, exosomal LRRK2 protein, RNA species, inflammatory cytokines, mitochondrial DNA (mtDNA), and intermediary metabolites. Pteridines, α-synuclein, mtDNA, 5-hydroxyindolacetic acid, β-D-glucose, lamp2, interleukin-8, and vascular endothelial growth factor were suggested to differentiate patients with LRRK2-related PD from sporadic PD patients; 8-hydroxy-2’-deoxyguanosine (8-OHdG), 8-isoprostane (8-ISO), 2-hydroxybutyrate, mtDNA, lamp2, and neopterin may differentiate between LRRK2 controls and LRRK2-related PD subjects; and soluble oligomeric α-synuclein, 8-OHdG, and 8-ISO might differentiate LRRK2 controls from unaffected subjects [101,120-127]. However, the low numbers of investigations of each analyte, small sample sizes, and methodological differences limit conclusions that can be drawn from these studies. Further investigations are indicated to determine the validity of the analytes identified in these studies as possible biomarkers for LRRK2 PD patients and to obtain markers for PD phenoconversion.
- Longitudinal studies in LRRK2-related PD families have shown fundamentally different neuropathological findings in phenotypically identical family members [72]. In a postmortem study with 38 patients with LRRK2-related PD with proven pathogenic LRRK2 mutations, 28 of the patients had symptoms compatible with typical levodopa-responsive parkinsonism, and neuropathological findings in 20 of the subjects were similar to those typically found in patients with idiopathic PD, with substantia nigra LBs. Nigral degeneration without LBs was found in 12 patients, diffuse LB disease was present in two subjects, tau pathology was found in two subjects, and α-synuclein-immunoreactive glial inclusions and ubiquitin-immunoreactive structures were found in one subject each. Similar findings with and without LBs in LRRK2-related PD have been reported by other investigators [128]. The range of neuropathological findings in LRRK2 PD might decrease the likelihood that α-synuclein oligomer, amyloid or tau concentrations in cerebrospinal fluid (CSF) or peripheral body fluids could offer biomarkers for LRRK2-related PD diagnosis or progression.
- The most striking difference between groups of LRRK2 mutation carriers is seen after DBS treatment. LRRK2-related PD patients develop levodopa-induced fluctuations and dyskinesias as patients do in the sporadic PD groups. The percentage of LRRK2 mutation carriers among those qualifying for DBS surgery is significantly higher than that among patients with sporadic PD [129]. The outcome from DBS surgery seems to depend on the location of the LRRK2 mutation at the gene. There have been a number of studies demonstrating superior efficiency and good outcome after DBS surgery in LRRK2 G2019S mutation carriers [129-132]. There are only a few cases published on patients carrying mutations in the ROC domain [78,133,134]. The results from these studies are all negative. Patients with LRRK2-related PD with mutations in the ROC domain seem to have less optimal outcome than those carrying mutations in the kinase domain. This might explain why there are so few reports after DBS in patients with LRRK2 ROC-domain mutations. There is a tendency not to report negative results and vice versa. There is only one report on DBS in patients with mutations in the LRRK2 COR domain [135]. These patients seem to experience a good outcome after DBS, comparable to that generally seen in G2019S patients.
- The long-term prognosis of LRRK2 mutation carriers might reflect the underlying neuropathology among the different mutations, which is quite heterogeneous [72,128,136]. Approximately half of LRRK2-related PD patients show no LB pathology, although the highest percentage is found in G2019S mutation carriers [128]. This is in some ways a paradox since the G2019S patients have the most favorable outcome after DBS, they have the best scores on smell tests and they seem to have the lowest rate of progression measured by annual increase in UPDRS scores [114,137,138].
- Autosomal-dominant mutations within the LRRK2 gene account for a small percentage of all cases of PD and a much higher proportion in some populations in the Middle East/North Africa. LRRK2-associated PD closely resembles idiopathic disease in terms of late age of onset, signs, and symptoms. The penetrance of LRRK2 mutations is incomplete and dependent on age and each specific mutation. Up to half of the LRRK2 patients do not show any LBs at autopsy.
- Long-term follow-up indicates that carriers of the most common mutation, G2019S, have slightly better prognosis than those with sporadic PD without any known mutation. Patients carrying the G2019S mutation selected for deep brain stimulation, DBS, seem to have very good long-term outcomes compared to cases with LRRK2 mutations in the ROC domain. The latter group should probably not be selected as good candidates for DBS.
- PARK17, VPS35-related PD
- The VPS35 gene was first discovered in a large Swiss family with multiple members affected by PD. At the same time, a group in Austria had similar cases with the D620N (p.Asp620Asn, c.1858G>A) mutation in the vacuolar protein sorting 35, VPS35, gene [139,140]. To date, there are only very few cases and families with this mutation published.
- The VPS35 phenotype varies, but in the few cases published to date, most of them have tremor-dominant PD with some tendency toward depression and other neuropsychiatric features. Members from one large Taiwanese family presented with a classic PD phenotype responding well to levodopa [141].
- Other polymorphisms in the VPS35 gene have since been identified, but these variants still need to be definitively linked to PD. VPS35 was originally identified in yeast as a member of the retromer complex. This complex is involved in the intracellular trafficking of proteins, where it may interact with parkin and may be involved in the retromer-mediated endosomal sorting process [142].
- There have been no systematic follow-up studies in patients with VPS35-related PD, with only a few single reports [143,144].
- One member of the Taiwanese family with VPS35-related PD had STN-DBS, and the outcome was reported as very successful [142].
- GBA mutations and variants causing PD
- It has been noted for many years that there is an increased frequency of parkinsonism among patients with GD (Table 2), and that there is an increased frequency of typical parkinsonism among healthy heterozygous mutation carriers of GBA gene mutations [8]. GD is a lysosomal storage disorder and results from the deficiency of the lysosomal enzyme glucocerebrosidase, GCase, localized to lysosomes. It is an autosomal-recessive disorder caused by mutations in the human β-glucocerebrosidase gene, GBA. Low GCase levels lead to the accumulation of its major substrate, glucosylceramide, GL-1, localized in lysosomes. In GD, large amounts of GL-1 accumulate in many cell types, including macrophages and neurons, which may lead to white matter accumulation, cell death and neurodegeneration [11,145].
- The GBA gene is located on chromosome 1q21 and comprises 11 exons and 10 introns, spanning 7.6 kb of sequence. A nonprocessed pseudogene (GBAP) that shares 96% exonic sequence homology is located 16 kb downstream of the functional GBA gene. The presence of this highly homologous pseudogene along with another 6 genes at the locus increases the occurrence of chromosomal rearrangements and misalignments in this region. These processes provide an explanation for the high number of complex recombinant alleles that have been detected in patients with GD [146].
- Mutations in the gene for GD are probably the most common familial variant of PD. The age-specific estimates of PD penetrance in patients with GD and GBA heterozygous carriers is still debated. Although GBA mutations may increase the risk for PD, the vast majority of patients with GD who are heterozygotes may not develop the disease [147-149]. Studies are ongoing to identify what modifies the risk of PD in GBA mutation carriers.
- In a French study, approximately 10% of PD patients were carriers of some GD mutation. PD was strongly associated with the neuropathic GD mutation and to a lesser degree to the more common GD variants [150]. The latter are correlated with PD, but they do not cause GD in homozygous carriers. In a large multicenter study, it was shown that the prevalence of GBA mutations strongly varies between ethnic groups [13].
- GBA patients are usually grouped on the basis of their mutations as having benign variants or mild or severe mutations. The most common variants are E326K, T369M and T297S. N370S is a milder mutation than L444P, which is regarded as a severe mutation. In addition, there are a high number of rare variants/mutations, which makes the genetic testing for GBA parkinsonism technically complicated and the interpretation of the results difficult. Future diagnostics for GBA mutations may be of importance for neuroprotective therapy. These biomarkers could include CSF glucocerebrosidase activity or some other CSF or plasma levels of glucosylceramide substrates.
- The mean age of onset of GBA-related PD is to some degree related to the type of mutation. The onset in neuropathic mutation carriers is approximately 50 years and is 5–10 years later in patients with the more benign N370S mutation. Patients carrying one of the GBA variants have an age of onset comparable to those carrying the N370S mutation.
- The phenotype is often quite typical and clinically not easy to separate from that seen in patients with sporadic PD, with resting tremor, bradykinesia, rigidity and postural instability in patients with advanced cases. Nonmotor features include cognitive impairment, hallucinations, autonomic dysfunction and sleep disorders. GBA PD patients typically have severe anosmia [151]. Most GBA-related PD patients show a good response to levodopa, and the development of levodopa-related complications such as dyskinesias and off-periods is probably comparable to that seen in sporadic PD patients. The progression of patients with GBA-related PD is quite fast; therefore, many patients may be candidates for DBS treatment. This may not always be a good choice since studies have shown that patients with GBArelated PD deteriorate faster and develop cognitive deficits after fewer years than patients with sporadic PD [132,151].
- PD in the context of GD seems to resemble idiopathic PD more closely in terms of its clinical heterogeneity than PD associated with GBA heterozygous mutations [152]. Heterozygous GBA mutation carriers develop a more severe phenotype, including dyskinesias, than those with sporadic PD [153,154].
- There have been several studies in GBA-related PD and the risk of developing dementia. Three studies investigated the effects of GBA variation on PD dementia, Parkinson’s disease dementia (PDD), and risk. Four addressed the effects of GBA mutations on PDD risk, two explored the effects of GBA polymorphism on PDD risk, two investigated the effects of the GBA L444P mutation on PDD risk, and two studied the effects of the GBA N370S mutation on the risk of cognitive decline. These studies came from different ethnic groups located in Europe and North America, and one was from Japan. One study predicted cognitive decline and dementia in half of the patients after 8–10 years of disease duration [150,155]. They made an algorithm, and the main indicators were age, education, depression and UPDRS [155]. Most studies indicate that PD patients carrying a GBA mutation seem to have a 3–5 times higher risk of developing dementia than noncarriers [154,156,157].
- Well-preserved cognitive function is also a crucial factor for survival in PD. After the discovery of the connection between GBA mutations and PD pathology, many reports have described cortical and/or hippocampal LBs in the majority of the subjects with GBA mutations, corresponding to the more aggressive parkinsonism described in probands with both GD and parkinsonism [158-160]. PD patients with GD-associated mutations develop dementia and psychosis significantly earlier than those without mutations, but wearing-off does not significantly differ from that observed in patients with mutation [154]. The risk for dementia is strongly modulated by the type of mutation. In the clinical continuum between PD and DLB, patients with GBA mutations seem to localize midway, with carriers of severe mutations closer to DLB than to idiopathic PD [157].
- The difference in the risk of developing so-called sporadic PD and GBA-related PD is particularly important in regard to the treatment of late-stage complications. The selection of patients for advanced PD therapy, including deep brain stimulation, is based on international consensus [161]. PD patients with GD-associated mutations develop wearing-off and dyskinesia following the same timeline as sporadic PD patients [154]. Many patients with GBA-related PD are thus, in principle, excellent candidates for DBS surgery. However, the shorter time to cognitive deterioration after DBS makes these patients less optimal for advanced treatment. In retrospect, many patients who have developed dementia and other neuropsychiatric complications not long after DBS have been shown to be GBA mutation carriers [48,131,132,162,163].
- GBA patients exhibit classic symptoms, including tremor, rigidity and bradykinesia. There are 2 relatively common GBA mutations, N370S and L444P, and there are a high number of rare mutations and variants. GBA mutations are most common among Ashkenazi Jewish PD patients, 15%, compared to as low as 3% of PD patients of other ethnic origin. Low levels of GCase may promote the stabilization of α-synuclein into oligomers similar to those found in LBs. This may explain why GBS patients have an earlier onset of PD symptoms with faster cognitive and motor decline than sporadic PD patients. GBA-associated PD patients present with more rapid disease progression of motor impairment, cognitive decline and lower survival rates than PD patients without mutations.
- Other rare mutations may cause PD but are not included in this review. These include mutations in EIF4G1 (eukaryotic translation initiation factor 4 gamma 1), DCTN1 (dynactin 1) in Perry syndrome, ATP13A2 (ATPase type 13A2), PARK9 in Kufor-Rakeb syndrome, PLA2G6 (phospholipase A2, group 6) (PARK14), and FBXO7 (F-box protein 7) (PARK15). There are no follow-up reports on these patients.
METHODS
Long-term outcome of SNCA-related PD
Summary
Long-term outcome of PRKN-related PD
Summary
Long-term outcome of PINK1-related PD
Summary
Long-term outcome of DJ-1-related PD
Summary
Long-term outcome of LRRK2-related PD
Summary
Long-term prognosis of GBA-related PD
Summary
Mutations not covered in this review
- During the last twenty years, there has been continued success in the identification of genetic causes and contributors to PD. There are now a high number of mutations and gene variants that may affect the risk for developing PD; the majority are very rare. This review describes the long-term outcome in PD patients carrying the most common mutations. The genotypephenotype relations that vary between different genetic mutations have provided excellent information for selecting patients for specific therapies. SNCA mutation carriers have a younger onset than LRRK2 mutation carriers and have a more severe prognosis. The early-onset recessive patients respond well to levodopa and are less prone to cognitive decline. The GBA-related PD phenotype is similar to that seen in sporadic PD, but patients with GBA-related PD have more nonmotor symptoms, including cognitive impairment and hallucinations, making them less optimal candidates for advanced treatments such as DBS.
- The clinical and basic research in genetic PD has given increasing insight into the genetic and molecular basis of this complex disease. This is a prerequisite for revealing the molecular pathogenesis of PD and identifying a therapy based on etiology.
CONCLUSIONS
- None.
Acknowledgments
Figure 1.The initial pedigree, presenting six kindreds traced back to a common founder couple, born in approximately 1,580 on a small island off the coast of Central Norway. Later, five more families were identified, more or less intermarried with the others. Adapted from Johansen et al. Parkinsonism Relat Disord 2010;16: 527-530.[93] The affected cases are abridged to maintain confidentiality. Later, five more families were identified, and genealogical links to the other LRRK2 families were found.

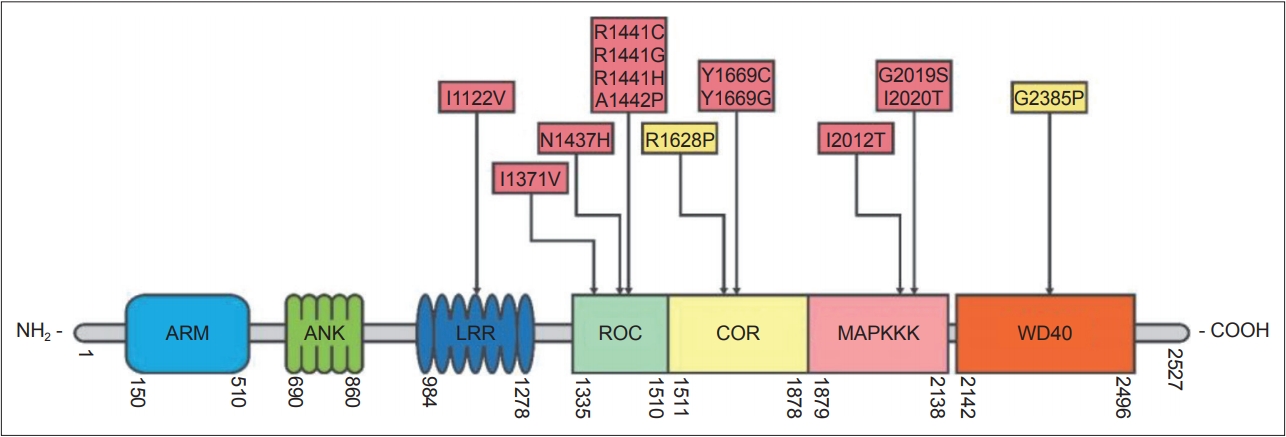
Table 1.Most common types of genetic Parkinson’s disease
Table 2.
GBA mutations and variants causing Parkinson’s disease
- 1. Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997;276:2045–2047.ArticlePubMed
- 2. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature 1997;388:839–840.ArticlePubMedPDF
- 3. Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998;392:605–608.ArticlePubMedPDF
- 4. Valente EM, Bentivoglio AR, Dixon PH, Ferraris A, Ialongo T, Frontali M, et al. Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am J Hum Genet 2001;68:895–900.ArticlePubMedPMC
- 5. Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003;299:256–259.ArticlePubMed
- 6. Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, van der Brug M, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004;44:595–600.ArticlePubMed
- 7. Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004;44:601–607.ArticlePubMed
- 8. Neudorfer O, Giladi N, Elstein D, Abrahamov A, Turezkite T, Aghai E, et al. Occurrence of Parkinson’s syndrome in type I Gaucher disease. QJM 1996;89:691–694.ArticlePubMedPDF
- 9. Chang D, Nalls MA, Hallgrímsdóttir IB, Hunkapiller J, van der Brug M, Cai F, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 2017;49:1511–1516.ArticlePubMedPMCPDF
- 10. Lunati A, Lesage S, Brice A. The genetic landscape of Parkinson’s disease. Rev Neurol (Paris) 2018;174:628–643.ArticlePubMed
- 11. Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C, et al. A review of gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci 2017;18:441.ArticlePubMedPMC
- 12. Mitsui J, Mizuta I, Toyoda A, Ashida R, Takahashi Y, Goto J, et al. Mutations for Gaucher disease confer high susceptibility to Parkinson disease. Arch Neurol 2009;66:571–576.ArticlePubMed
- 13. Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 2009;361:1651–1661.ArticlePubMedPMC
- 14. Hoehn MM, Yahr MD. Parkinsonism: onset, progression and mortality. Neurology 1967;17:427–442.ArticlePubMed
- 15. Curtis L, Lees AJ, Stern GM, Marmot MG. Effect of L-dopa on course of Parkinson’s disease. Lancet 1984;2:211–212.ArticlePubMed
- 16. Hely MA, Morris JG, Reid WG, Trafficante R. Sydney Multicenter Study of Parkinson’s disease: non-L-dopa-responsive problems dominate at 15 years. Mov Disord 2005;20:190–199.ArticlePubMed
- 17. Vollstedt EJ, Kasten M, Klein C; MJFF Global Genetic Parkinson’s Disease Study Group. Using global team science to identify genetic parkinson’s disease worldwide. Ann Neurol 2019;86:153–157.ArticlePubMedPMC
- 18. Golbe LI, Di Iorio G, Sanges G, Lazzarini AM, La Sala S, Bonavita V, et al. Clinical genetic analysis of Parkinson’s disease in the Contursi kindred. Ann Neurol 1996;40:767–775.ArticlePubMed
- 19. Trinh J, Zeldenrust FMJ, Huang J, Kasten M, Schaake S, Petkovic S, et al. Genotype-phenotype relations for the Parkinson’s disease genes SNCA, LRRK2, VPS35: MDSGene systematic review. Mov Disord 2018;33:1857–1870.ArticlePubMed
- 20. Spellman GG. Report of familial cases of parkinsonism. Evidence of a dominant trait in a patient’s family. JAMA 1962;179:372–374.ArticlePubMed
- 21. Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 2004;55:164–173.ArticlePubMed
- 22. Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, Weir D, et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord 2013;28:811–813.ArticlePubMed
- 23. Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 1998;18:106–108.ArticlePubMedPDF
- 24. Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, et al. Novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol Aging 2014;35:2180.e1–2180.e5.ArticlePubMed
- 25. Elia AE, Petrucci S, Fasano A, Guidi M, Valbonesi S, Bernardini L, et al. Alpha-synuclein gene duplication: marked intrafamilial variability in two novel pedigrees. Mov Disord 2013;28:813–817.ArticlePubMed
- 26. Fuchs J, Nilsson C, Kachergus J, Munz M, Larsson EM, Schüle B, et al. Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology 2007;68:916–922.ArticlePubMed
- 27. Ross OA, Braithwaite AT, Skipper LM, Kachergus J, Hulihan MM, Middleton FA, et al. Genomic investigation of alpha-synuclein multiplication and parkinsonism. Ann Neurol 2008;63:743–750.ArticlePubMed
- 28. Ricciardi L, Petrucci S, Di Giuda D, Serra L, Spanò B, Sensi M, et al. The Contursi family 20 years later: intrafamilial phenotypic variability of the SNCA p.A53T mutation. Mov Disord 2016;31:257–258.ArticlePubMed
- 29. Swerdlow RH, Parks JK, Cassarino DS, Binder DR, Bennett JP Jr, Di Iorio G, et al. Biochemical analysis of cybrids expressing mitochondrial DNA from Contursi kindred Parkinson’s subjects. Exp Neurol 2001;169:479–485.ArticlePubMed
- 30. Muenter MD, Forno LS, Hornykiewicz O, Kish SJ, Maraganore DM, Caselli RJ, et al. Hereditary form of parkinsonism--dementia. Ann Neurol 1998;43:768–781.ArticlePubMed
- 31. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. α-Synuclein locus triplication causes Parkinson’s disease. Science 2003;302:841.ArticlePubMed
- 32. Picillo M, Lizarraga KJ, Friesen EL, Chau H, Zhang M, Sato C, et al. Parkinsonism due to A53E α-synuclein gene mutation: clinical, genetic, epigenetic, and biochemical features. Mov Disord 2018;33:1950–1955.ArticlePubMed
- 33. Martikainen MH, Päivärinta M, Hietala M, Kaasinen V. Clinical and imaging findings in Parkinson disease associated with the A53E SNCA mutation. Neurol Genet 2015;1:e27.ArticlePubMedPMC
- 34. Antonini A, Pilleri M, Padoan A, Landi A, Ferla S, Biundo R, et al. Successful subthalamic stimulation in genetic Parkinson’s disease caused by duplication of the α-synuclein gene. J Neurol 2012;259:165–167.ArticlePubMedPDF
- 35. Cartelli D, Amadeo A, Calogero AM, Casagrande FVM, De Gregorio C, Gioria M, et al. Parkin absence accelerates microtubule aging in dopaminergic neurons. Neurobiol Aging 2018;61:66–74.ArticlePubMed
- 36. Johansen KK, Torp SH, Farrer MJ, Gustavsson EK, Aasly JO. A Case of Parkinson’s disease with no Lewy body pathology due to a homozygous exon deletion in Parkin. Case Rep Neurol Med 2018;2018:6838965.ArticlePubMedPMCPDF
- 37. Ahlskog JE. Parkin and PINK1 parkinsonism may represent nigral mitochondrial cytopathies distinct from Lewy body Parkinson’s disease. Parkinsonism Relat Disord 2009;15:721–727.ArticlePubMedPMC
- 38. Aasly JO, Sæther O, Johansen KK, Bathen TF, Giskeødegård GF, White LR. Changes to intermediary metabolites in sporadic and LRRK2 Parkinson’s disease demonstrated by proton magnetic resonance spectroscopy. Parkinsons Dis 2015;2015:264896.ArticlePubMedPMCPDF
- 39. Mellick GD, Siebert GA, Funayama M, Buchanan DD, Li Y, Imamichi Y, et al. Screening PARK genes for mutations in early-onset Parkinson’s disease patients from Queensland, Australia. Parkinsonism Relat Disord 2009;15:105–109.ArticlePubMed
- 40. Weissbach A, König IR, Hückelheim K, Pramstaller PP, Werner E, Brüggemann N, et al. Influence of L-dopa on subtle motor signs in heterozygous Parkin- and PINK1 mutation carriers. Parkinsonism Relat Disord 2017;42:95–99.ArticlePubMed
- 41. Clarimon J, Johnson J, Djaldetti R, Hernandez D, Hattori N, Sroka H, et al. Mutation of the Parkin gene in a Persian family: clinical progression over a 40-year period. Mov Disord 2005;20:887–890.ArticlePubMed
- 42. Doherty KM, Silveira-Moriyama L, Parkkinen L, Healy DG, Farrell M, Mencacci NE, et al. Parkin disease: a clinicopathologic entity? JAMA Neurol 2013;70:571–579.ArticlePubMedPMC
- 43. Morgante F, Fasano A, Ginevrino M, Petrucci S, Ricciardi L, Bove F, et al. Impulsive-compulsive behaviors in parkin-associated Parkinson disease. Neurology 2016;87:1436–1441.ArticlePubMedPMC
- 44. Srivastava A, Tang MX, Mejia-Santana H, Rosado L, Louis ED, Caccappolo E, et al. The relation between depression and parkin genotype: the CORE-PD study. Parkinsonism Relat Disord 2011;17:740–744.ArticlePubMedPMC
- 45. Kim HJ, Yun JY, Kim YE, Lee JY, Kim HJ, Kim JY, et al. Parkin mutation and deep brain stimulation outcome. J Clin Neurosci 2014;21:107–110.ArticlePubMed
- 46. Moro E, Volkmann J, König IR, Winkler S, Hiller A, Hassin-Baer S, et al. Bilateral subthalamic stimulation in Parkin and PINK1 parkinsonism. Neurology 2008;70:1186–1191.ArticlePubMed
- 47. Romito LM, Contarino MF, Ghezzi D, Franzini A, Garavaglia B, Albanese A. High frequency stimulation of the subthalamic nucleus is efficacious in Parkin disease. J Neurol 2005;252:208–211.ArticlePubMedPDF
- 48. Rizzone MG, Martone T, Balestrino R, Lopiano L. Genetic background and outcome of Deep Brain Stimulation in Parkinson’s disease. Parkinsonism Relat Disord 2019;64:8–19.ArticlePubMed
- 49. Doherty KM, Hardy J. Parkin disease and the Lewy body conundrum. Mov Disord 2013;28:702–704.ArticlePubMedPMC
- 50. Kasten M, Hartmann C, Hampf J, Schaake S, Westenberger A, Vollstedt EJ, et al. Genotype-phenotype relations for the Parkinson’s disease genes Parkin, PINK1, DJ1: MDSGene systematic review. Mov Disord 2018;33:730–741.ArticlePubMed
- 51. Bonifati V, Rohé CF, Breedveld GJ, Fabrizio E, De Mari M, Tassorelli C, et al. Early-onset parkinsonism associated with PINK1 mutations: frequency, genotypes, and phenotypes. Neurology 2005;65:87–95.ArticlePubMed
- 52. Toft M, Myhre R, Pielsticker L, White LR, Aasly JO, Farrer MJ. PINK1 mutation heterozygosity and the risk of Parkinson’s disease. J Neurol Neurosurg Psychiatry 2007;78:82–84.ArticlePubMedPMC
- 53. Ishihara-Paul L, Hulihan MM, Kachergus J, Upmanyu R, Warren L, Amouri R, et al. PINK1 mutations and parkinsonism. Neurology 2008;71:896–902.ArticlePubMedPMC
- 54. Ricciardi L, Petrucci S, Guidubaldi A, Ialongo T, Serra L, Ferraris A, et al. Phenotypic variability of PINK1 expression: 12 years’ clinical follow-up of two Italian families. Mov Disord 2014;29:1561–1566.ArticlePubMed
- 55. Ferraris A, Ialongo T, Passali GC, Pellecchia MT, Brusa L, Laruffa M, et al. Olfactory dysfunction in Parkinsonism caused by PINK1 mutations. Mov Disord 2009;24:2350–2357.ArticlePubMed
- 56. Kertelge L, Brüggemann N, Schmidt A, Tadic V, Wisse C, Dankert S, et al. Impaired sense of smell and color discrimination in monogenic and idiopathic Parkinson’s disease. Mov Disord 2010;25:2665–2669.ArticlePubMed
- 57. Borellini L, Cogiamanian F, Carrabba G, Locatelli M, Rampini P, Di Fonzo A, et al. Globus pallidus internus deep brain stimulation in PINK-1 related Parkinson’s disease: a case report. Parkinsonism Relat Disord 2017;38:93–94.ArticlePubMed
- 58. Samaranch L, Lorenzo-Betancor O, Arbelo JM, Ferrer I, Lorenzo E, Irigoyen J, et al. PINK1-linked parkinsonism is associated with Lewy body pathology. Brain 2010;133:1128–1142.ArticlePubMedPDF
- 59. Steele JC, Guella I, Szu-Tu C, Lin MK, Thompson C, Evans DM, et al. Defining neurodegeneration on Guam by targeted genomic sequencing. Ann Neurol 2015;77:458–468.ArticlePubMed
- 60. Bonifati V. Autosomal recessive parkinsonism. Parkinsonism Relat Disord 2012;18 Suppl 1:S4–S6.ArticlePubMed
- 61. Clark LN, Afridi S, Mejia-Santana H, Harris J, Louis ED, Cote LJ, et al. Analysis of an early-onset Parkinson’s disease cohort for DJ-1 mutations. Mov Disord 2004;19:796–800.ArticlePubMed
- 62. Klein C, Westenberger A. Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med 2012;2:a008888.ArticlePubMedPMC
- 63. Gustavsson EK, Trinh J, McKenzie M, Bortnick S, Petersen MS, Farrer MJ, et al. Genetic identification in early onset parkinsonism among Norwegian patients. Mov Disord Clin Pract 2017;4:499–508.ArticlePubMedPMC
- 64. Bozi M, Papadimitriou D, Antonellou R, Moraitou M, Maniati M, Vassilatis DK, et al. Genetic assessment of familial and early-onset Parkinson’s disease in a Greek population. Eur J Neurol 2014;21:963–968.ArticlePubMed
- 65. García S, López-Hernández LB, Suarez-Cuenca JA, Solano-Rojas M, Gallegos-Arreola MP, Gama-Moreno O, et al. Low prevalence of most frequent pathogenic variants of six PARK genes in sporadic Parkinson’s disease. Folia Neuropathol 2014;52:22–29.ArticlePubMed
- 66. Alcalay RN, Caccappolo E, Mejia-Santana H, Tang MX, Rosado L, Ross BM, et al. Frequency of known mutations in early-onset Parkinson disease: implication for genetic counseling: the consortium on risk for early onset Parkinson disease study. Arch Neurol 2010;67:1116–1122.ArticlePubMedPMC
- 67. Taipa R, Pereira C, Reis I, Alonso I, Bastos-Lima A, Melo-Pires M, et al. DJ-1 linked parkinsonism (PARK7) is associated with Lewy body pathology. Brain 2016;139(Pt 6):1680–1687.ArticlePubMedPDF
- 68. Abbas MM, Govindappa ST, Sudhaman S, Thelma BK, Juyal RC, Behari M, et al. Early onset Parkinson’s disease due to DJ1 mutations: an Indian study. Parkinsonism Relat Disord 2016;32:20–24.ArticlePubMed
- 69. Narendra DP, Isonaka R, Nguyen D, Schindler AB, Kokkinis AD, Ehrlich D, et al. Peripheral synucleinopathy in a DJ1 patient with Parkinson disease, cataracts, and hearing loss. Neurology 2019;92:1113–1115.ArticlePubMedPMC
- 70. Kuusimäki T, Korpela J, Pekkonen E, Martikainen MH, Antonini A, Kaasinen V. Deep brain stimulation for monogenic Parkinson’s disease: a systematic review. J Neurol 2020;267:883–897.ArticlePubMedPDF
- 71. Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F. A new locus for Parkinson’s disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann Neurol 2002;51:296–301.ArticlePubMed
- 72. Wszolek ZK, Pfeiffer RF, Tsuboi Y, Uitti RJ, McComb RD, Stoessl AJ, et al. Autosomal dominant parkinsonism associated with variable synuclein and tau pathology. Neurology 2004;62:1619–1622.ArticlePubMed
- 73. Kachergus J, Mata IF, Hulihan M, Taylor JP, Lincoln S, Aasly J, et al. Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am J Hum Genet 2005;76:672–680.ArticlePubMedPMC
- 74. Aasly JO, Toft M, Fernandez-Mata I, Kachergus J, Hulihan M, White LR, et al. Clinical features of LRRK2-associated Parkinson’s disease in central Norway. Ann Neurol 2005;57:762–765.ArticlePubMed
- 75. Funayama M, Hasegawa K, Ohta E, Kawashima N, Komiyama M, Kowa H, et al. An LRRK2 mutation as a cause for the parkinsonism in the original PARK8 family. Ann Neurol 2005;57:918–921.ArticlePubMed
- 76. Rideout HJ, Stefanis L. The neurobiology of LRRK2 and its role in the pathogenesis of Parkinson’s disease. Neurochem Res 2014;39:576–592.ArticlePubMedPDF
- 77. Shu L, Zhang Y, Pan H, Xu Q, Guo J, Tang B, et al. Clinical Heterogeneity Among LRRK2 Variants in Parkinson’s Disease: A Meta-Analysis. Front Aging Neurosci 2018;10:283.ArticlePubMedPMC
- 78. Aasly JO, Vilariño-Güell C, Dachsel JC, Webber PJ, West AB, Haugarvoll K, et al. Novel Pathogenic Lrrk2 p.Asn1437His substitution in familial Parkinson’s disease. Mov Disord 2010;25:2156–2163.ArticlePubMedPMC
- 79. Cook DA, Kannarkat GT, Cintron AF, Butkovich LM, Fraser KB, Chang J, et al. LRRK2 levels in immune cells are increased in Parkinson’s disease. NPJ Parkinsons Dis 2017;3:11.ArticlePubMedPMCPDF
- 80. Shu L, Zhang Y, Sun Q, Pan H, Tang B. A comprehensive analysis of population differences in LRRK2 variant distribution in Parkinson’s disease. Front Aging Neurosci 2019;11:13.ArticlePubMedPMC
- 81. Farrer MJ, Stone JT, Lin CH, Dächsel JC, Hulihan MM, Haugarvoll K, et al. Lrrk2 G2385R is an ancestral risk factor for Parkinson’s disease in Asia. Parkinsonism Relat Disord 2007;13:89–92.ArticlePubMed
- 82. Liang D, Shu L, Pan H, Xu Q, Guo J, Yan X, et al. Clinical characteristics of PD patients with LRRK2 G2385R and R1628P variants. Neurosci Lett 2018;685:185–189.ArticlePubMed
- 83. Kelly K, Wang S, Boddu R, Liu Z, Moukha-Chafiq O, Augelli-Szafran C, et al. The G2019S mutation in LRRK2 imparts resiliency to kinase inhibition. Exp Neurol 2018;309:1–13.ArticlePubMedPMC
- 84. Ozelius LJ, Senthil G, Saunders-Pullman R, Ohmann E, Deligtisch A, Tagliati M, et al. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N Engl J Med 2006;354:424–425.ArticlePubMed
- 85. Orr-Urtreger A, Shifrin C, Rozovski U, Rosner S, Bercovich D, Gurevich T, et al. The LRRK2 G2019S mutation in Ashkenazi Jews with Parkinson disease: is there a gender effect? Neurology 2007;69:1595–1602.ArticlePubMed
- 86. Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol 2008;7:583–590.ArticlePubMedPMC
- 87. Clark LN, Wang Y, Karlins E, Saito L, Mejia-Santana H, Harris J, et al. Frequency of LRRK2 mutations in early- and late-onset Parkinson disease. Neurology 2006;67:1786–1791.ArticlePubMed
- 88. Latourelle JC, Sun M, Lew MF, Suchowersky O, Klein C, Golbe LI, et al. The Gly2019Ser mutation in LRRK2 is not fully penetrant in familial Parkinson’s disease: the GenePD study. BMC Med 2008;6:32.ArticlePubMedPMCPDF
- 89. Tan EK, Tang M, Tan LC, Wu YR, Wu RM, Ross OA, et al. Lrrk2 R1628P in non-Chinese Asian races. Ann Neurol 2008;64:472–473.ArticlePubMed
- 90. Ross OA, Wu YR, Lee MC, Funayama M, Chen ML, Soto AI, et al. Analysis of Lrrk2 R1628P as a risk factor for Parkinson’s disease. Ann Neurol 2008;64:88–92.ArticlePubMed
- 91. Rudenko IN, Kaganovich A, Hauser DN, Beylina A, Chia R, Ding J, et al. The G2385R variant of leucine-rich repeat kinase 2 associated with Parkinson’s disease is a partial loss-of-function mutation. Biochem J 2012;446:99–111.ArticlePubMedPMCPDF
- 92. Rudenko IN, Kaganovich A, Langston RG, Beilina A, Ndukwe K, Kumaran R, et al. The G2385R risk factor for Parkinson’s disease enhances CHIP-dependent intracellular degradation of LRRK2. Biochem J 2017;474:1547–1558.ArticlePubMedPMCPDF
- 93. Johansen KK, Hasselberg K, White LR, Farrer MJ, Aasly JO. Genealogical studies in LRRK2-associated Parkinson’s disease in central Norway. Parkinsonism Relat Disord 2010;16:527–530.ArticlePubMed
- 94. Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 2014;46:989–993.ArticlePubMedPMCPDF
- 95. Ross OA, Soto-Ortolaza AI, Heckman MG, Aasly JO, Abahuni N, Annesi G, et al. Association of LRRK2 exonic variants with susceptibility to Parkinson’s disease: a case-control study. Lancet Neurol 2011;10:898–908.ArticlePubMedPMC
- 96. Yuan X, Chen Y, Cao B, Zhao B, Wei Q, Guo X, et al. An association analysis of the R1628P and G2385R polymorphisms of the LRRK2 gene in multiple system atrophy in a Chinese population. Parkinsonism Relat Disord 2015;21:147–149.ArticlePubMed
- 97. Cho HJ, Liu G, Jin SM, Parisiadou L, Xie C, Yu J, et al. MicroRNA-205 regulates the expression of Parkinson’s disease-related leucine-rich repeat kinase 2 protein. Hum Mol Genet 2013;22:608–620.ArticlePubMedPDF
- 98. Bliederhaeuser C, Zondler L, Grozdanov V, Ruf WP, Brenner D, Melrose HL, et al. LRRK2 contributes to monocyte dysregulation in Parkinson’s disease. Acta Neuropathol Commun 2016;4:123.ArticlePubMedPMCPDF
- 99. Di Maio R, Hoffman EK, Rocha EM, Keeney MT, Sanders LH, De Miranda BR, et al. LRRK2 activation in idiopathic Parkinson’s disease. Sci Transl Med 2018;10:eaar5429.ArticlePubMedPMC
- 100. Fraser KB, Rawlins AB, Clark RG, Alcalay RN, Standaert DG, Liu N, et al. Ser(P)-1292 LRRK2 in urinary exosomes is elevated in idiopathic Parkinson’s disease. Mov Disord 2016;31:1543–1550.ArticlePubMedPMC
- 101. Wang S, Liu Z, Ye T, Mabrouk OS, Maltbie T, Aasly J, et al. Elevated LRRK2 autophosphorylation in brain-derived and peripheral exosomes in LRRK2 mutation carriers. Acta Neuropathol Commun 2017;5:86.ArticlePubMedPMCPDF
- 102. Roosen DA, Cookson MR. LRRK2 at the interface of autophagosomes, endosomes and lysosomes. Mol Neurodegener 2016;11:73.ArticlePubMedPMCPDF
- 103. Wang S, West AB. Caught in the act: LRRK2 in exosomes. Biochem Soc Trans 2019;47:663–670.ArticlePubMedPMCPDF
- 104. Zhang M, Yao C, Cai J, Liu S, Liu XN, Chen Y, et al. LRRK2 is involved in the pathogenesis of system lupus erythematosus through promoting pathogenic antibody production. J Transl Med 2019;17:37.ArticlePubMedPMCPDF
- 105. Cresto N, Gardier C, Gubinelli F, Gaillard MC, Liot G, West AB, et al. The unlikely partnership between LRRK2 and α-synuclein in Parkinson’s disease. Eur J Neurosci 2019;49:339–363.ArticlePubMed
- 106. Daher JP, Volpicelli-Daley LA, Blackburn JP, Moehle MS, West AB. Abrogation of α-synuclein-mediated dopaminergic neurodegeneration in LRRK2-deficient rats. Proc Natl Acad Sci U S A 2014;111:9289–9294.ArticlePubMedPMC
- 107. Daher JP, Abdelmotilib HA, Hu X, Volpicelli-Daley LA, Moehle MS, Fraser KB, et al. Leucine-rich Repeat Kinase 2 (LRRK2) Pharmacological Inhibition Abates α-Synuclein Gene-induced Neurodegeneration. J Biol Chem 2015;290:19433–19444.ArticlePubMedPMC
- 108. Nucifora FC Jr, Nucifora LG, Ng CH, Arbez N, Guo Y, Roby E, et al. Ubiqutination via K27 and K29 chains signals aggregation and neuronal protection of LRRK2 by WSB1. Nat Commun 2016;7:11792.ArticlePubMedPMCPDF
- 109. Sossi V, de la Fuente-Fernández R, Nandhagopal R, Schulzer M, McKenzie J, Ruth TJ, et al. Dopamine turnover increases in asymptomatic LRRK2 mutations carriers. Mov Disord 2010;25:2717–2723.ArticlePubMed
- 110. Wile DJ, Agarwal PA, Schulzer M, Mak E, Dinelle K, Shahinfard E, et al. Serotonin and dopamine transporter PET changes in the premotor phase of LRRK2 parkinsonism: cross-sectional studies. Lancet Neurol 2017;16:351–359.ArticlePubMedPMC
- 111. Liu SY, Wile DJ, Fu JF, Valerio J, Shahinfard E, McCormick S, et al. The effect of LRRK2 mutations on the cholinergic system in manifest and premanifest stages of Parkinson’s disease: a cross-sectional PET study. Lancet Neurol 2018;17:309–316.ArticlePubMedPMC
- 112. Gaig C, Vilas D, Infante J, Sierra M, García-Gorostiaga I, Buongiorno M, et al. Nonmotor symptoms in LRRK2 G2019S associated Parkinson’s disease. PLoS One 2014;9:e108982.ArticlePubMedPMC
- 113. Johansen KK, Warø BJ, Aasly JO. Olfactory dysfunction in sporadic Parkinson’s Disease and LRRK2 carriers. Acta Neurol Scand 2014;129:300–306.ArticlePubMed
- 114. Saunders-Pullman R, Mirelman A, Alcalay RN, Wang C, Ortega RA, Raymond D, et al. Progression in the LRRK2-asssociated Parkinson disease population. JAMA Neurol 2018;75:312–319.ArticlePubMedPMC
- 115. Alcalay RN, Mirelman A, Saunders-Pullman R, Tang MX, Mejia Santana H, Raymond D, et al. Parkinson disease phenotype in Ashkenazi Jews with and without LRRK2 G2019S mutations. Mov Disord 2013;28:1966–1971.ArticlePubMed
- 116. Yahalom G, Orlev Y, Cohen OS, Kozlova E, Friedman E, Inzelberg R, et al. Motor progression of Parkinson’s disease with the leucine-rich repeat kinase 2 G2019S mutation. Mov Disord 2014;29:1057–1060.ArticlePubMed
- 117. Hentati F, Trinh J, Thompson C, Nosova E, Farrer MJ, Aasly JO. LRRK2 parkinsonism in Tunisia and Norway: a comparative analysis of disease penetrance. Neurology 2014;83:568–569.ArticlePubMedPMC
- 118. Trinh J, Gustavsson EK, Vilariño-Güell C, Bortnick S, Latourelle J, McKenzie MB, et al. DNM3 and genetic modifiers of age of onset in LRRK2 Gly2019Ser parkinsonism: a genome-wide linkage and association study. Lancet Neurol 2016;15:1248–1256.ArticlePubMed
- 119. Oosterveld LP, Allen JC Jr, Ng EY, Seah SH, Tay KY, Au WL, et al. Greater motor progression in patients with Parkinson disease who carry LRRK2 risk variants. Neurology 2015;85:1039–1042.ArticlePubMed
- 120. Klaver AC, Coffey MP, Aasly JO, Loeffler DA. CSF lamp2 concentrations are decreased in female Parkinson’s disease patients with LRRK2 mutations. Brain Res 2018;1683:12–16.ArticlePubMed
- 121. Shi M, Furay AR, Sossi V, Aasly JO, Armaly J, Wang Y, et al. DJ-1 and αSYN in LRRK2 CSF do not correlate with striatal dopaminergic function. Neurobiol Aging 2012;33:836.e5–836.e7.ArticlePubMed
- 122. Aasly JO, Johansen KK, Brønstad G, Warø BJ, Majbour NK, Varghese S, et al. Elevated levels of cerebrospinal fluid α-synuclein oligomers in healthy asymptomatic LRRK2 mutation carriers. Front Aging Neurosci 2014;6:248.ArticlePubMedPMC
- 123. Vilas D, Shaw LM, Taylor P, Berg D, Brockmann K, Aasly J, et al. Cerebrospinal fluid biomarkers and clinical features in leucine-rich repeat kinase 2 (LRRK2) mutation carriers. Mov Disord 2016;31:906–914.ArticlePubMed
- 124. Loeffler DA, Klaver AC, Coffey MP, Aasly JO, LeWitt PA. Increased oxidative stress markers in cerebrospinal fluid from healthy subjects with Parkinson’s Disease-associated LRRK2 gene mutations. Front Aging Neurosci 2017;9:89.ArticlePubMedPMC
- 125. Ichinose H, Inoue KI, Arakawa S, Watanabe Y, Kurosaki H, Koshiba S, et al. Alterations in the reduced pteridine contents in the cerebrospinal fluids of LRRK2 mutation carriers and patients with Parkinson’s disease. J Neural Transm (Vienna) 2018;125:45–52.ArticlePubMedPDF
- 126. Aasly JO, Shi M, Sossi V, Stewart T, Johansen KK, Wszolek ZK, et al. Cerebrospinal fluid amyloid β and tau in LRRK2 mutation carriers. Neurology 2012;78:55–61.ArticlePubMedPMC
- 127. Podlesniy P, Vilas D, Taylor P, Shaw LM, Tolosa E, Trullas R. Mitochondrial DNA in CSF distinguishes LRRK2 from idiopathic Parkinson’s disease. Neurobiol Dis 2016;94:10–17.ArticlePubMed
- 128. Kalia LV, Lang AE, Hazrati LN, Fujioka S, Wszolek ZK, Dickson DW, et al. Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol 2015;72:100–105.ArticlePubMedPMC
- 129. Johansen KK, Jørgensen JV, White LR, Farrer MJ, Aasly JO. Parkinsonrelated genetics in patients treated with deep brain stimulation. Acta Neurol Scand 2011;123:201–206.ArticlePubMed
- 130. Schüpbach M, Lohmann E, Anheim M, Lesage S, Czernecki V, Yaici S, et al. Subthalamic nucleus stimulation is efficacious in patients with Parkinsonism and LRRK2 mutations. Mov Disord 2007;22:119–122.ArticlePubMed
- 131. Pal GD, Hall D, Ouyang B, Phelps J, Alcalay R, Pauciulo MW, et al. Genetic and clinical predictors of deep brain stimulation in young-onset Parkinson’s disease. Mov Disord Clin Pract 2016;3:465–471.ArticlePubMedPMC
- 132. Angeli A, Mencacci NE, Duran R, Aviles-Olmos I, Kefalopoulou Z, Candelario J, et al. Genotype and phenotype in Parkinson’s disease: lessons in heterogeneity from deep brain stimulation. Mov Disord 2013;28:1370–1375.ArticlePubMedPMC
- 133. Gómez-Esteban JC, Lezcano E, Zarranz JJ, González C, Bilbao G, Lambarri I, et al. Outcome of bilateral deep brain subthalamic stimulation in patients carrying the R1441G mutation in the LRRK2 dardarin gene. Neurosurgery 2008;62:857–862.ArticlePubMedPDF
- 134. Hatano T, Funayama M, Kubo SI, Mata IF, Oji Y, Mori A, et al. Identification of a Japanese family with LRRK2 p.R1441G-related Parkinson’s disease. Neurobiol Aging 2014;35:2656.e17–2656.e23.ArticlePubMed
- 135. Perju-Dumbrava LD, McDonald M, Kneebone AC, Long R, Thyagarajan D. Sustained response to deep brain stimulation in LRRK2 parkinsonism with the Y1699C mutation. J Parkinsons Dis 2012;2:269–271.ArticlePubMed
- 136. Compta Y, Parkkinen L, O’Sullivan SS, Vandrovcova J, Holton JL, Collins C, et al. Lewy- and Alzheimer-type pathologies in Parkinson’s disease dementia: which is more important? Brain 2011;134(Pt 5):1493–1505.ArticlePubMedPMCPDF
- 137. Thaler A, Kozlovski T, Gurevich T, Bar-Shira A, Gana-Weisz M, Orr-Urtreger A, et al. Survival rates among Parkinson’s disease patients who carry mutations in the LRRK2 and GBA genes. Mov Disord 2018;33:1656–1660.ArticlePubMed
- 138. Clarimón J, Pagonabarraga J, Paisán-Ruíz C, Campolongo A, Pascual-Sedano B, Martí-Massó JF, et al. Tremor dominant parkinsonism: clinical description and LRRK2 mutation screening. Mov Disord 2008;23:518–523.ArticlePubMed
- 139. Vilariño-Güell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet 2011;89:162–167.ArticlePubMedPMC
- 140. Zimprich A, Benet-Pagès A, Struhal W, Graf E, Eck SH, Offman MN, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet 2011;89:168–175.ArticlePubMedPMC
- 141. Chen YF, Chang YY, Lan MY, Chen PL, Lin CH. Identification of VPS35 p.D620N mutation-related Parkinson’s disease in a Taiwanese family with successful bilateral subthalamic nucleus deep brain stimulation: a case report and literature review. BMC Neurol 2017;17:191.ArticlePubMedPMCPDF
- 142. Williams ET, Glauser L, Tsika E, Jiang H, Islam S, Moore DJ. Parkin mediates the ubiquitination of VPS35 and modulates retromer-dependent endosomal sorting. Hum Mol Genet 2018;27:3189–3205.ArticlePubMedPMCPDF
- 143. Sheerin UM, Charlesworth G, Bras J, Guerreiro R, Bhatia K, Foltynie T, et al. Screening for VPS35 mutations in Parkinson’s disease. Neurobiol Aging 2012;33:838.e1–838.e5.ArticlePubMed
- 144. Kumar KR, Weissbach A, Heldmann M, Kasten M, Tunc S, Sue CM, et al. Frequency of the D620N mutation in VPS35 in Parkinson disease. Arch Neurol 2012;69:1360–1364.ArticlePubMed
- 145. Romero R, Ramanathan A, Yuen T, Bhowmik D, Mathew M, Munshi LB, et al. Mechanism of glucocerebrosidase activation and dysfunction in Gaucher disease unraveled by molecular dynamics and deep learning. Proc Natl Acad Sci U S A 2019;116:5086–5095.ArticlePubMedPMC
- 146. Martínez-Arias R, Comas D, Mateu E, Bertranpetit J. Glucocerebrosidase pseudogene variation and Gaucher disease: recognizing pseudogene tracts in GBA alleles. Hum Mutat 2001;17:191–198.ArticlePubMed
- 147. Rana HQ, Balwani M, Bier L, Alcalay RN. Age-specific Parkinson disease risk in GBA mutation carriers: information for genetic counseling. Genet Med 2013;15:146–149.ArticlePubMedPDF
- 148. Alcalay RN, Dinur T, Quinn T, Sakanaka K, Levy O, Waters C, et al. Comparison of Parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol 2014;71:752–757.ArticlePubMedPMC
- 149. Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol 2012;11:986–998.ArticlePubMedPMC
- 150. Liu G, Boot B, Locascio JJ, Jansen IE, Winder-Rhodes S, Eberly S, et al. Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann Neurol 2016;80:674–685.ArticlePubMedPMC
- 151. McNeill A, Duran R, Proukakis C, Bras J, Hughes D, Mehta A, et al. Hyposmia and cognitive impairment in Gaucher disease patients and carriers. Mov Disord 2012;27:526–532.ArticlePubMedPMC
- 152. Collins LM, Williams-Gray CH, Morris E, Deegan P, Cox TM, Barker RA. The motor and cognitive features of Parkinson’s disease in patients with concurrent Gaucher disease over 2 years: a case series. J Neurol 2018;265:1789–1794.ArticlePubMedPMCPDF
- 153. Kim HJ, Mason S, Foltynie T, Winder-Rhodes S, Barker RA, Williams-Gray CH. Motor complications in Parkinson’s disease: 13-year follow-up of the CamPaIGN cohort. Mov Disord 2020;35:185–190.ArticlePubMed
- 154. Oeda T, Umemura A, Mori Y, Tomita S, Kohsaka M, Park K, et al. Impact of glucocerebrosidase mutations on motor and nonmotor complications in Parkinson’s disease. Neurobiol Aging 2015;36:3306–3313.ArticlePubMed
- 155. Liu G, Locascio JJ, Corvol JC, Boot B, Liao Z, Page K, et al. Prediction of cognition in Parkinson’s disease with a clinical-genetic score: a longi tudinal analysis of nine cohorts. Lancet Neurol 2017;16:620–629.ArticlePubMedPMC
- 156. Winder-Rhodes SE, Evans JR, Ban M, Mason SL, Williams-Gray CH, Foltynie T, et al. Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain 2013;136(Pt 2):392–399.ArticlePubMedPDF
- 157. Cilia R, Tunesi S, Marotta G, Cereda E, Siri C, Tesei S, et al. Survival and dementia in GBA-associated Parkinson’s disease: the mutation matters. Ann Neurol 2016;80:662–673.ArticlePubMed
- 158. Lwin A, Orvisky E, Goker-Alpan O, LaMarca ME, Sidransky E. Glucocerebrosidase mutations in subjects with parkinsonism. Mol Genet Metab 2004;81:70–73.ArticlePubMed
- 159. Brockmann K, Srulijes K, Pflederer S, Hauser AK, Schulte C, Maetzler W, et al. GBA-associated Parkinson’s disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov Disord 2015;30:407–411.ArticlePubMed
- 160. Pal G, Robertson E, O’Keefe J, Hall D. The neuropsychiatric and motor profile of GBA-associated Parkinson’s disease: a review. Mov Disord Clin Pract 2015;3:4–8.ArticlePubMedPMC
- 161. Munhoz RP, Picillo M, Fox SH, Bruno V, Panisset M, Honey CR, et al. Eligibility criteria for deep brain stimulation in Parkinson’s disease, tremor, and dystonia. Can J Neurol Sci 2016;43:462–471.ArticlePubMed
- 162. Lythe V, Athauda D, Foley J, Mencacci NE, Jahanshahi M, Cipolotti L, et al. GBA-associated Parkinson’s disease: progression in a deep brain stimulation cohort. J Parkinsons Dis 2017;7:635–644.ArticlePubMed
- 163. Singleton AB, Farrer MJ, Bonifati V. The genetics of Parkinson’s disease: progress and therapeutic implications. Mov Disord 2013;28:14–23.ArticlePubMedPMC
REFERENCES
Figure & Data
References
Citations
Citations to this article as recorded by 
- Navigating the Frontiers of Machine Learning in Neurodegenerative Disease Therapeutics
Yoonjeong Cha, Mohamedi N. Kagalwala, Jermaine Ross
Pharmaceuticals.2024; 17(2): 158. CrossRef - The Molecular Impact of Glucosylceramidase Beta 1 (Gba1) in Parkinson’s Disease: a New Genetic State of the Art
Júlio César Claudino dos Santos, Gabriela Braga Cabrera Mano, André Rodrigues da Cunha Barreto-Vianna, Tulia Fernanda Meira Garcia, Aline Vieira de Vasconcelos, Caio Sérgio Gomes Sá, Sarah Lopes de Souza Santana, Ana Gabriela Ponte Farias, Beatriz Seimaru
Molecular Neurobiology.2024;[Epub] CrossRef - Early Onset Parkinson’s Disease and It’s Genetic Consequences
Hatice ÖMERCİKOĞLU ÖZDEN, Dilek GÜNAL
OSMANGAZİ JOURNAL OF MEDICINE.2024;[Epub] CrossRef - Genotype–phenotype correlation in PRKN-associated Parkinson’s disease
Poornima Jayadev Menon, Sara Sambin, Baptiste Criniere-Boizet, Thomas Courtin, Christelle Tesson, Fanny Casse, Melanie Ferrien, Louise-Laure Mariani, Stephanie Carvalho, Francois-Xavier Lejeune, Sana Rebbah, Gaspard Martet, Marion Houot, Aymeric Lanore, G
npj Parkinson's Disease.2024;[Epub] CrossRef - Factors Influencing Patient Disclosure of Parkinson's Disease Genetic Testing Results to Relatives
Jeanine Schulze, Jasmine Kaur Dhaliwal, Mandy Miller, Emily Quinn, Leah Wetherill, Lola Cook
Movement Disorders Clinical Practice.2024;[Epub] CrossRef - Staging Parkinson’s Disease According to the MNCD (Motor/Non-motor/Cognition/Dependency) Classification Correlates with Disease Severity and Quality of Life
Diego Santos-García, Teresa de Deus Fonticoba, Carlos Cores Bartolomé, Maria J. Feal Painceiras, Maria Cristina Íñiguez-Alvarado, Iago García Díaz, Silvia Jesús, Maria Teresa Buongiorno, Lluís Planellas, Marina Cosgaya, Juan García Caldentey, Nuria Caball
Journal of Parkinson's Disease.2023; 13(3): 379. CrossRef - To lump or to split? Deep brain stimulation may improve non-motor symptoms in certain Parkinson's disease subtypes
Neepa Patel
Parkinsonism & Related Disorders.2023; 109: 105369. CrossRef - Emerging Roles of Signal Transduction Pathways in Neurodegenerative Diseases. Hunting New Possible Therapeutic Molecular Targets
Vincenza Rita Lo Vasco
OBM Geriatrics.2023; 07(02): 1. CrossRef - Genome- and Exome-Wide Association Studies Revealed Candidate Genes Associated with DaTscan Imaging Features
Arash Yaghoobi, Homa Seyedmirzaei, Moein Ala, Cristine Alves da Costa
Parkinson's Disease.2023; 2023: 1. CrossRef - Cardiac 123I-Metaiodobenzylguanidine (MIBG) Scintigraphy in Parkinson’s Disease: A Comprehensive Review
Jamir Pitton Rissardo, Ana Letícia Fornari Caprara
Brain Sciences.2023; 13(10): 1471. CrossRef - Applicability of clinical genetic testing for deep brain stimulation treatment in monogenic Parkinson’s disease and monogenic dystonia: a multidisciplinary team perspective
Valentino Rački, Mario Hero, Eliša Papić, Gloria Rožmarić, Nada Starčević Čizmarević, Darko Chudy, Borut Peterlin, Vladimira Vuletić
Frontiers in Neuroscience.2023;[Epub] CrossRef - PTEN-Induced Putative Kinase 1 Dysfunction Accelerates Synucleinopathy
Tinh Thi Nguyen, Yun Joong Kim, Thuy Thi Lai, Phuong Thi Nguyen, Young Ho Koh, Linh Thi Nhat Nguyen, Hyeo-il Ma, Young Eun Kim
Journal of Parkinson's Disease.2022; 12(4): 1201. CrossRef - Oncogenic Pathways in Neurodegenerative Diseases
Luis Varela, Maria E. R. Garcia-Rendueles
International Journal of Molecular Sciences.2022; 23(6): 3223. CrossRef - Genetic Determinants of Survival in Parkinson's Disease in the Asian Population
Chunyu Li, Yanbing Hou, Ruwei Ou, Xiaojing Gu, Yongping Chen, Lingyu Zhang, Kuncheng Liu, Junyu Lin, Bei Cao, Qianqian Wei, Xueping Chen, Wei Song, Bi Zhao, Ying Wu, Yiyuan Cui, Huifang Shang
Movement Disorders.2022; 37(8): 1624. CrossRef - Obituary for Jan O. Aasly (1950–2022)
Matthew J. Farrer
Movement Disorders.2022; 37(9): 1783. CrossRef - Genetics in parkinson’s disease: From better disease understanding to machine learning based precision medicine
Mohamed Aborageh, Peter Krawitz, Holger Fröhlich
Frontiers in Molecular Medicine.2022;[Epub] CrossRef - Recent developments in nucleic acid-based therapies for Parkinson’s disease: Current status, clinical potential, and future strategies
Shivam Kumar Pandey, Rakesh Kumar Singh
Frontiers in Pharmacology.2022;[Epub] CrossRef - Genetic stratification of motor and QoL outcomes in Parkinson's disease in the EARLYSTIM study
Daniel Weiss, Zied Landoulsi, Patrick May, Manu Sharma, Michael Schüpbach, Hana You, Jean Christophe Corvol, Steffen Paschen, Ann-Kristin Helmers, Michael Barbe, Gereon Fink, Andrea A. Kühn, Christine Brefel Courbon, Lars Wojtecki, Philippe Damier, Valeri
Parkinsonism & Related Disorders.2022; 103: 169. CrossRef - Inflammatory Diseases Among Norwegian LRRK2 Mutation Carriers. A 15-Years Follow-Up of a Cohort
Jan O. Aasly
Frontiers in Neuroscience.2021;[Epub] CrossRef - Increased Mortality in Young-Onset Parkinson’s Disease
Eldbjørg Hustad, Tor Åge Myklebust, Sasha Gulati, Jan O. Aasly
Journal of Movement Disorders.2021; 14(3): 214. CrossRef - Contributing Factors and Evolution of Impulse Control Disorder in the Luxembourg Parkinson Cohort
Sylvia Binck, Claire Pauly, Michel Vaillant, Geraldine Hipp, Manon Gantenbein, Rejko Krueger, Nico J Diederich
Frontiers in Neurology.2020;[Epub] CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite- Figure
-
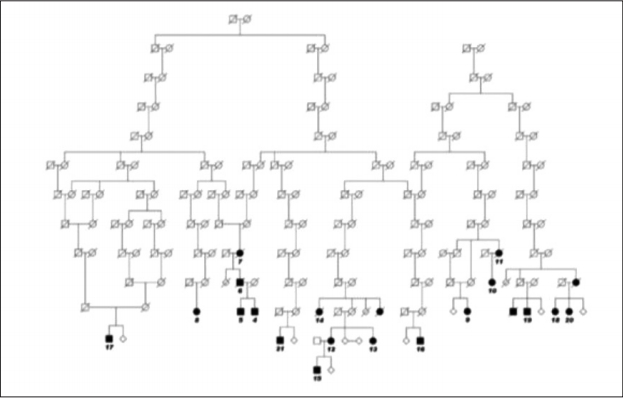
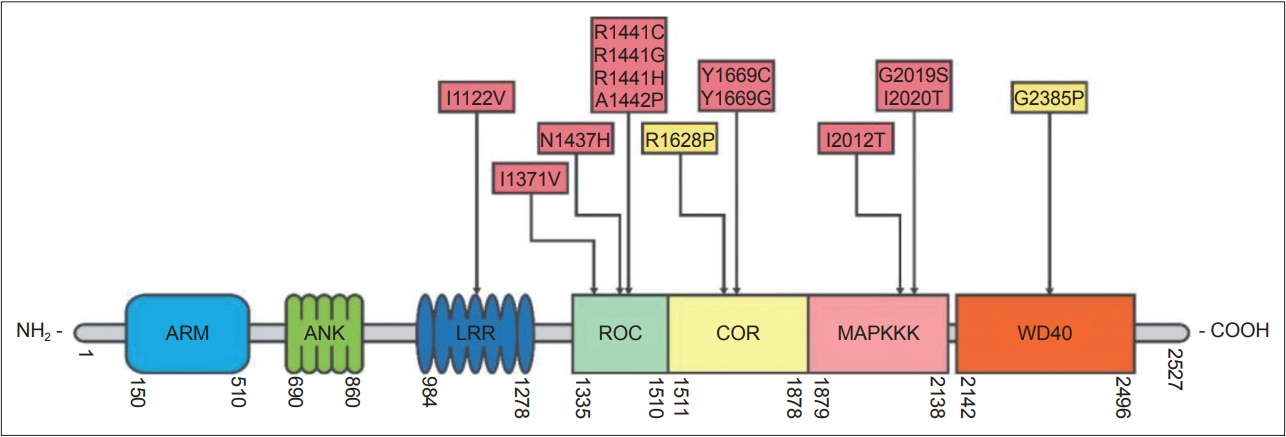
- Related articles
-
- A Survey of Perspectives on Telemedicine for Patients With Parkinson’s Disease
- Investigation of the Long-Term Effects of Amantadine Use in Parkinson’s Disease
- Mask on, Mask off: Subclinical Parkinson’s Disease Unveiled by COVID-19
- Hand Movement-Induced Eyeblink Bursts in a Patient With Parkinson’s Disease
- Umami and Other Taste Perceptions in Patients With Parkinson’s Disease
