 E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 13(3); 2020 > Article
-
Review Article
Emerging Concepts of Motor Reserve in Parkinson’s Disease -
Seok Jong Chung1,2
 , Jae Jung Lee3, Phil Hyu Lee1,4, Young H. Sohn1
, Jae Jung Lee3, Phil Hyu Lee1,4, Young H. Sohn1 -
Journal of Movement Disorders 2020;13(3):171-184.
DOI: https://doi.org/10.14802/jmd.20029
Published online: August 31, 2020
1Department of Neurology, Yonsei University College of Medicine, Seoul, Korea
2Department of Neurology, Yongin Severance Hospital, Yonsei University Health System, Yongin, Korea
3Department of Neurology, Ilsan Paik Hospital, Inje University College of Medicine, Goyang, Korea
4Severance Biomedical Science Institute, Yonsei University College of Medicine, Seoul, Korea
- Corresponding author: Young H. Sohn, MD, PhD Department of Neurology, Yonsei University College of Medicine, 50 Yonsei-ro, Seodaemun-gu, Seoul 03722, Korea / Tel: +82-2-2228-1601 / Fax: +82-2-393-0705 / E-mail: yhsohn62@yuhs.ac
Copyright © 2020 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- The concept of cognitive reserve (CR) in Alzheimer’s disease (AD) explains the differences between individuals in their susceptibility to AD-related pathologies. An enhanced CR may lead to less cognitive deficits despite severe pathological lesions. Parkinson’s disease (PD) is also a common neurodegenerative disease and is mainly characterized by motor dysfunction related to striatal dopaminergic depletion. The degree of motor deficits in PD is closely correlated to the degree of dopamine depletion; however, significant individual variations still exist. Therefore, we hypothesized that the presence of motor reserve (MR) in PD explains the individual differences in motor deficits despite similar levels of striatal dopamine depletion. Since 2015, we have performed a series of studies investigating MR in de novo patients with PD using the data of initial clinical presentation and dopamine transporter PET scan. In this review, we summarized the results of these published studies. In particular, some premorbid experiences (i.e., physical activity and education) and modifiable factors (i.e., body mass index and white matter hyperintensity on brain image studies) could modulate an individual’s capacity to tolerate PD pathology, which can be maintained throughout disease progression.
- Since 2009, dopamine transporter (DAT) scan using an [18F] N-(3-fluoropropyl)-2β-carbon ethoxy-3β-(4-iodophenyl) nortropane (FP-CIT) positron emission tomography (PET) scan was performed as an initial evaluation for the diagnosis of PD in Yonsei Parkinson Center. When patients were drug-naïve, Part III of Unified Parkinson’s Disease Rating Scale (UPDRS-motor) and the Mini-Mental State Examination (MMSE) were assessed in each patient to estimate their baseline PD motor severity and baseline cognitive function, respectively. The final diagnosis of PD was made according to the clinical criteria of the UK Brain Bank [6], the presence of appropriate DAT defects on FP-CIT PET scans [7], and the presence of PD drug response during follow-up (≥ 6 months). Patients with cognitive dysfunction (MMSE score < 24) were excluded from data analysis.
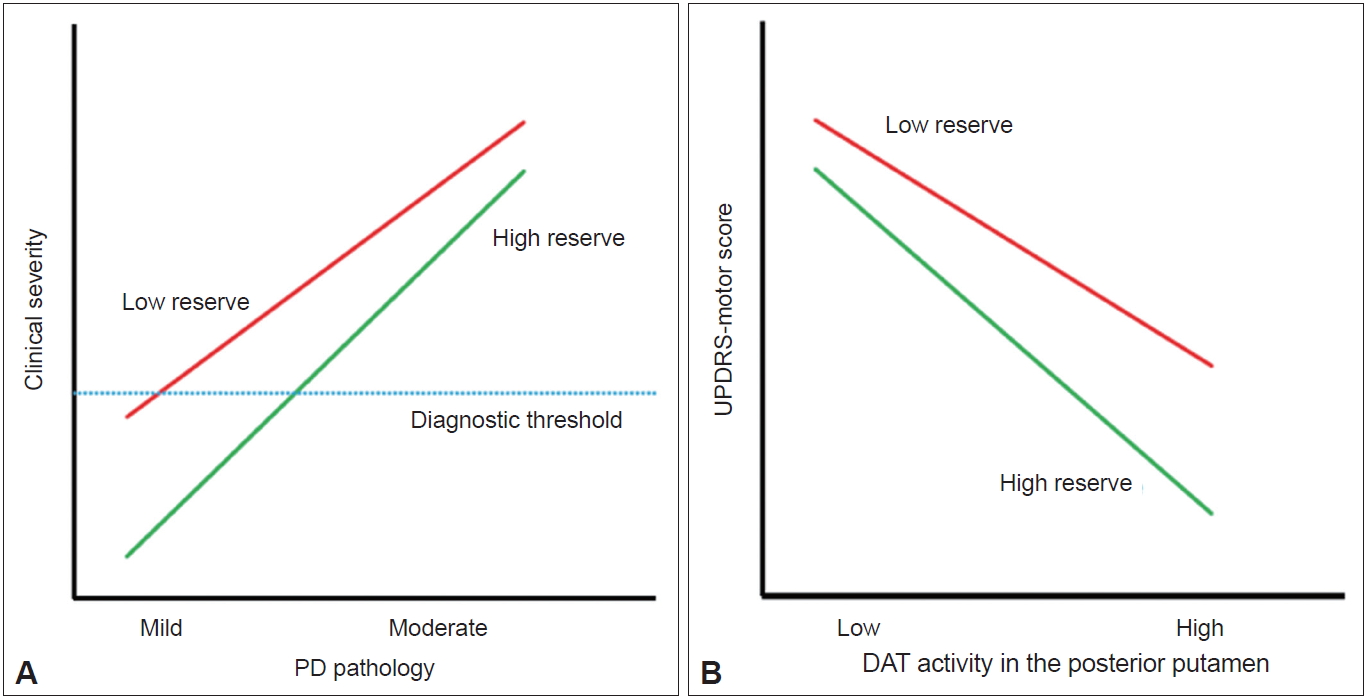
- The detailed methods of PET-CT image acquisition and quantitative analyses of FP-CIT PET data were described previously [8]. DAT activity was calculated in six striatal subregions: the ventral striatum, anterior caudate, posterior caudate, anterior putamen, ventral putamen and posterior putamen [7]. Baseline UPDRS motor score and DAT activity in the posterior putamen or in the sensorimotor striatum were used to assess MR in each patient with PD. The degree of CR in AD is represented by clinical severity and AD pathology [2]. If the same concept is applied to PD, MR may be represented by the initial UPDRS motor score (i.e., clinical severity) and the level of dopamine depletion (i.e., PD pathology) (Figure 1A). However, because we could not measure the level of dopamine depletion based on FP-CIT PET data, the X-axis of the graph should be replaced by DAT activity in the posterior putamen, and MR could be plotted as demonstrated in Figure 1B. In this figure, patients with high MR exhibited fewer motor deficits, i.e., lower UPDRS motor score, compared with those with low MR despite similar levels of DAT activity in the posterior putamen.
ASSESSMENT OF MR IN PD
- Exercise engagement
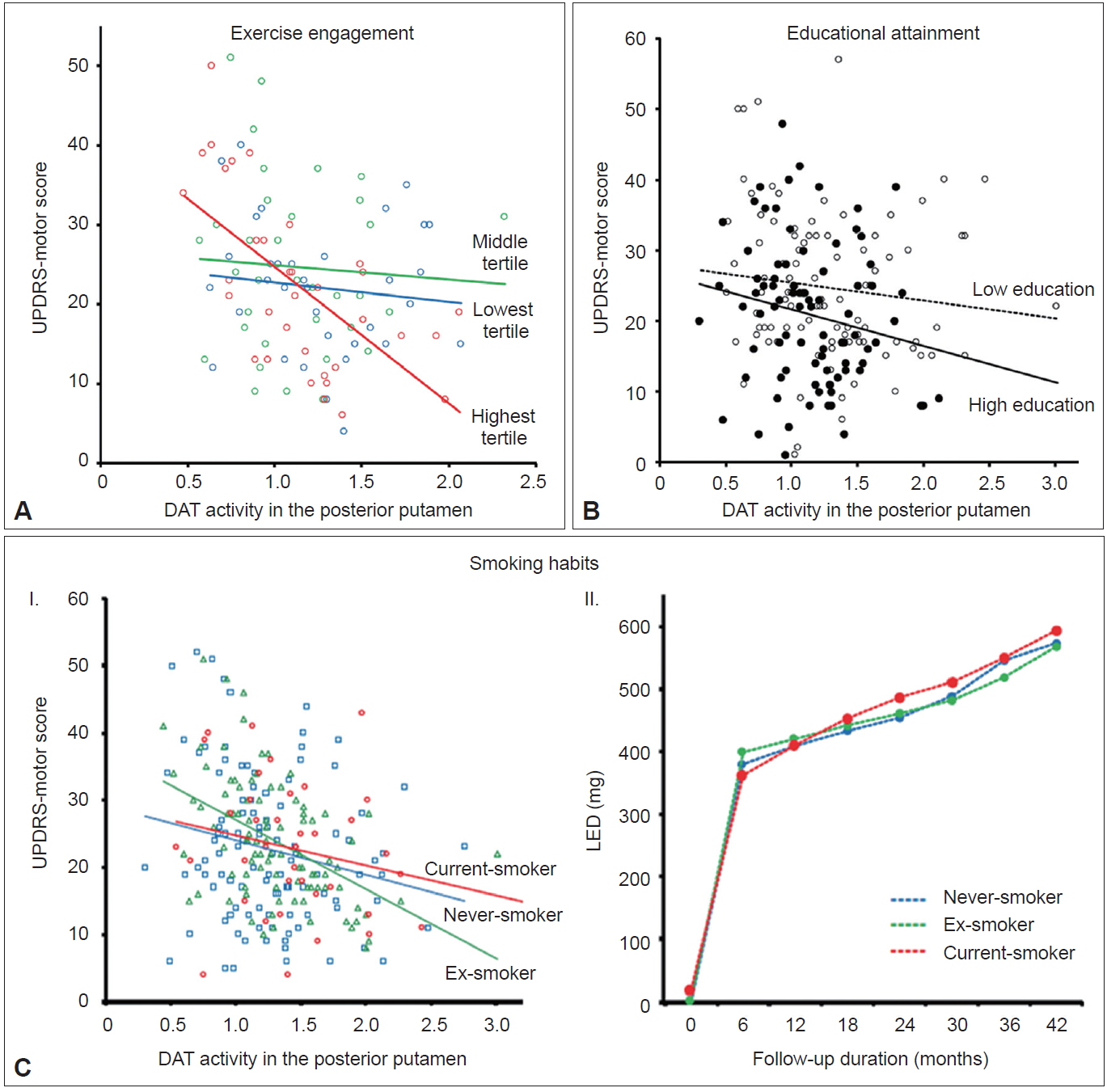
- Animal experiments have demonstrated that physical training may protect dopamine neurons from various parkinsonism-inducing neurotoxins [9,10]. Additionally, epidemiological studies have demonstrated that long-term strenuous exercises are associated with reduced development of PD in humans [11,12]. Accordingly, just as premorbid cognitive activity enhances an individual’s CR [2], premorbid physical activity (i.e., engagement in exercise) may enhance MR in individuals with PD. To address this hypothesis, premorbid engagement of leisure-time exercise was assessed in 102 patients using the Physical Activity Scale for the Elderly [13]. Among patients with mild to moderate reductions in striatal dopaminergic activity (greater than the median DAT activity in the posterior putamen), the group with the highest exercise engagement showed significantly lower UPDRS motor scores (i.e., less parkinsonian motor deficits) despite having similar levels of DAT activity compared to the middle and the lowest exercise engagement groups combined (Figure 2A). Meanwhile, among the patients with severe reduction in striatal dopaminergic activity, the highest tertile exercise group showed significantly higher UPDRS motor scores, suggesting a more rapid decline in motor function related to decreases in striatal DAT activity compared with the other two groups. These results suggest that engagement in premorbid exercise acts as a proxy for an active reserve in the motor domain (i.e., MR) in patients with PD. A recent paper demonstrated delayed clinical manifestation of PD among physically active skiers, which supports the above conclusion [14].
- Educational attainment
- Educational attainment is a main factor that enhances CR in AD [2]. In PD populations, an epidemiological study identified an inverse relationship between the level of education and the risk of PD [15]; however, others studies have reported inconsistent results [16,17]. Therefore, similar to CR in AD, higher educational attainment could also be associated with enhanced MR in PD. In this study, a total of 182 patients were classified into 2 groups: higher (≥ 12 years of education) and lower education (< 12 years of education) [18]. The higher education group exhibited less severe parkinsonian motor deficits and lower DAT activity in the posterior putamen than the lower education group despite a similar duration of PD symptoms. The difference in motor deficits between the groups remained significant after adjusting for potential confounding factors (i.e., age, sex, disease duration, and MMSE scores) as well as DAT activity in the posterior putamen as covariates (Figure 2B). These results suggest that high education attainment could lead to enhanced MR in individuals with PD. Educational attainment has also been proposed as an MR proxy in other previous studies [19-22], and a higher level of education possibly enhances MR via greater cerebral volume or white matter integrity, increased synaptic plasticity, more efficient recruitment of brain networks or recovery mechanisms [19,20], and beneficial effects on general health or environmental risk factors [21].
- Smoking habits
- Cigarette smoking is associated with a reduced risk of PD; a meta-analysis showed that current smokers have a relative risk of 0.39 for the development of PD [23]. However, whether smoking protects against the development of PD or PD itself protects against smoking remains controversial [24,25]. In experimental animals, cigarette smoke protects against toxin-induced dopamine neuronal damage [26,27]. However, it remains unknown whether smoking protects dopamine neuronal degeneration and subsequently enhances MR in humans with PD. Of the total 282 male patients with PD, current-smokers (n = 44) showed higher DAT activity in the posterior and ventral putamen but exhibited similar motor deficits compared to exsmokers (n = 105) and never-smokers (n = 133) [28]. Selective sparing of DAT activity in the posterior and ventral putamen was more pronounced in the more affected side than the less affected side, suggesting that smoking preferentially exerts a protective effect on dopamine neurons that are most affected by PD pathology. However, a similar slope of motor deficits relative to DAT activity in the posterior putamen suggests that smoking status is not associated with MR in individuals with PD (Figure 2C–I). Longitudinal change of levodopa-equivalent dose (LED) did not differ among the smoking groups (Figure 2C–II), which suggests no additional clinical benefits related to current smoking.
PREMORBID EXPERIENCES AND MR
- Age
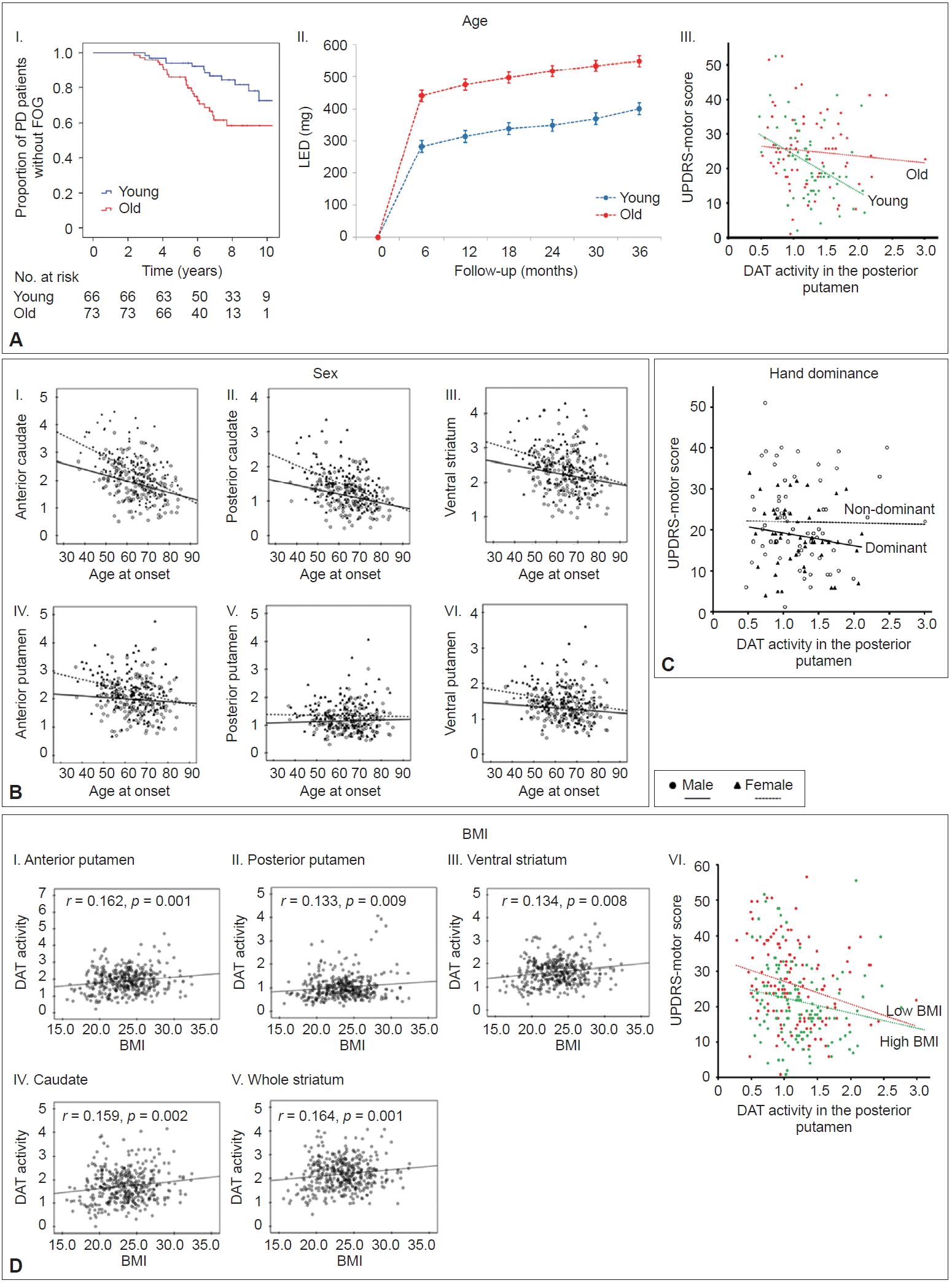
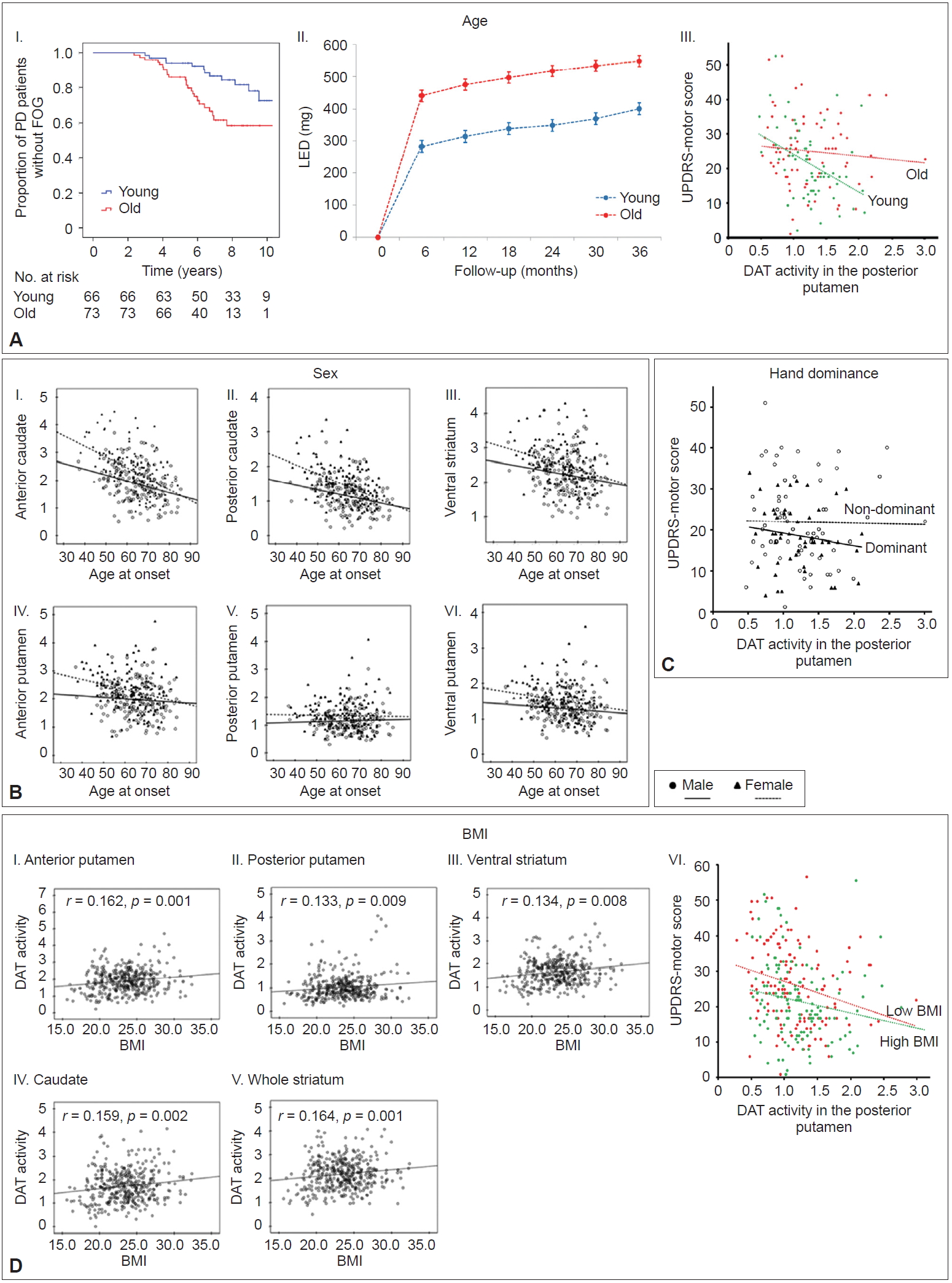
- Ample evidence suggests that age at PD onset is a major determinant of clinical heterogeneity in patients with PD [29-31]. Young-onset PD has consistently demonstrated slower disease progression, better response to dopaminergic medications, more frequent motor complications, and less frequent cognitive impairments compared to old-onset PD [31]. However, the mechanism underlying the age at onset-dependent differences in PD remains unknown. Few reports have demonstrated that age at onset is associated with different patterns of striatal dopamine depletion in PD [32-35]; however, these studies were limited by small sample size [33], confounding effects of PD medication [32,34], and less detailed segmentation of the striatum [32-35]. To investigate the relationship between age at onset and MR in PD, a total of 205 patients were subdivided into tertile groups according to their age at onset of PD symptoms [36]. The old-onset PD group (the highest tertile group, > 66 years, n = 73) exhibited higher UPDRS motor scores than the young-onset PD group (the lowest tertile group, < 58 years, n = 66), but DAT activity in the posterior putamen was comparable between the two groups. The old-onset PD group exhibited an increased risk for developing freezing of gait and required higher doses of dopaminergic medications for symptom control over time than the youngonset group (Figure 3A–I and II). In an additional analysis, a general linear model showed that the old-onset group had significantly more severe motor deficits than the young-onset group after controlling for sex, disease duration, and DAT activity in the posterior putamen (p < 0.05) (Figure 3A–III). This finding suggests that the old-onset group represents poor MR in PD compared to the young-onset group.
- Sex
- PD develops more often in males than in females; a meta-analysis showed an increased relative risk of 1.5 in males [37]. Age at PD onset is later in females than in males [38,39], which partly correlates with the fertile life span in females [39]. Clinically, female PD patients exhibit less severe parkinsonian motor features and better levodopa responses with more severe levodopa-induced dyskinesia [40,41]. However, whether sex is a critical risk factor for developing levodopa-induced dyskinesia remains controversial [42-44]. These sex differences suggest a beneficial influence of the female sex hormones (i.e., estrogen) against the development and progression of PD. The beneficial effect of estrogen could also enhance MR in individuals with PD, which may depend on the patients’ age. Among a total of 307 patients [45], female PD patients (n = 155) exhibited greater DAT activity in all the striatal subregions than male PD patients (n = 152). Age-related DAT decline was greater in the anterior and posterior caudate and in the anterior putamen in female PD patients compared to male PD patients but similar in other subregions (Figure 3B). Sex differences in age-related DAT decline in the antero-dorsal striatum is presumably due to age-related decline in estrogen. However, this difference was not observed in the sensorimotor striatum, suggesting that female PD patients do not have a greater MR than male PD patients.
- Hand dominance
- Handedness is the most prominent human behavioral asymmetry [46]. Compared to the nondominant primary motor cortex (M1), the dominant M1 exhibits a greater dispersion of elementary movement representations with more profuse horizontal connections [47-49]. Therefore, it is conceivable that the dominant hemisphere could have more efficient motor networks with a greater neural reserve to cope with the pathological changes related to PD. Unilateral onset and persistent asymmetry of motor signs are unique features of PD [50,51]. To evaluate whether dominant-side onset PD patients showed greater MR compared to nondominant-side onset PD patients, 118 PD patients with significant asymmetric motor deficits were included for analysis [52]. Among them, dominant-side onset patients (n = 57) exhibited fewer motor deficits despite exhibiting a similar level of DAT activity in the posterior putamen than the nondominant-side onset patients (n = 61) (Figure 3C), which suggests greater MR in dominant-side onset patients.
- Body mass index
- Patients with PD have a lower body mass index (BMI) than healthy subjects, which can be traced back almost 10 years before the diagnosis of PD [53]. In addition, a low BMI in PD is more pronounced in patients with greater disease severity [54]. Therefore, a low BMI could be associated with PD-related pathology (i.e., dopamine neuronal degeneration). A total of 398 patients were divided into five quintile groups according to their BMI [55]. This study demonstrated that BMI was associated with DAT activity in all striatal subregions (Figure 3D–I, II, III, IV, and V), which suggests that a lower BMI might be related to a lower density of nigrostriatal dopaminergic neurons in PD. In an additional analysis, we compared MR between the low BMI group (the first and second quintile group) and the high BMI group (the fourth and fifth quintile group). This analysis showed that the low BMI group exhibited greater motor deficits than the high BMI group after controlling for age, sex, and DAT activity in the posterior putamen (Figure 3D–VI). This result suggests that a low BMI in patients with early PD may represent low MR.
DEMOGRAPHIC VARIABLES AND MR
- Olfactory dysfunction
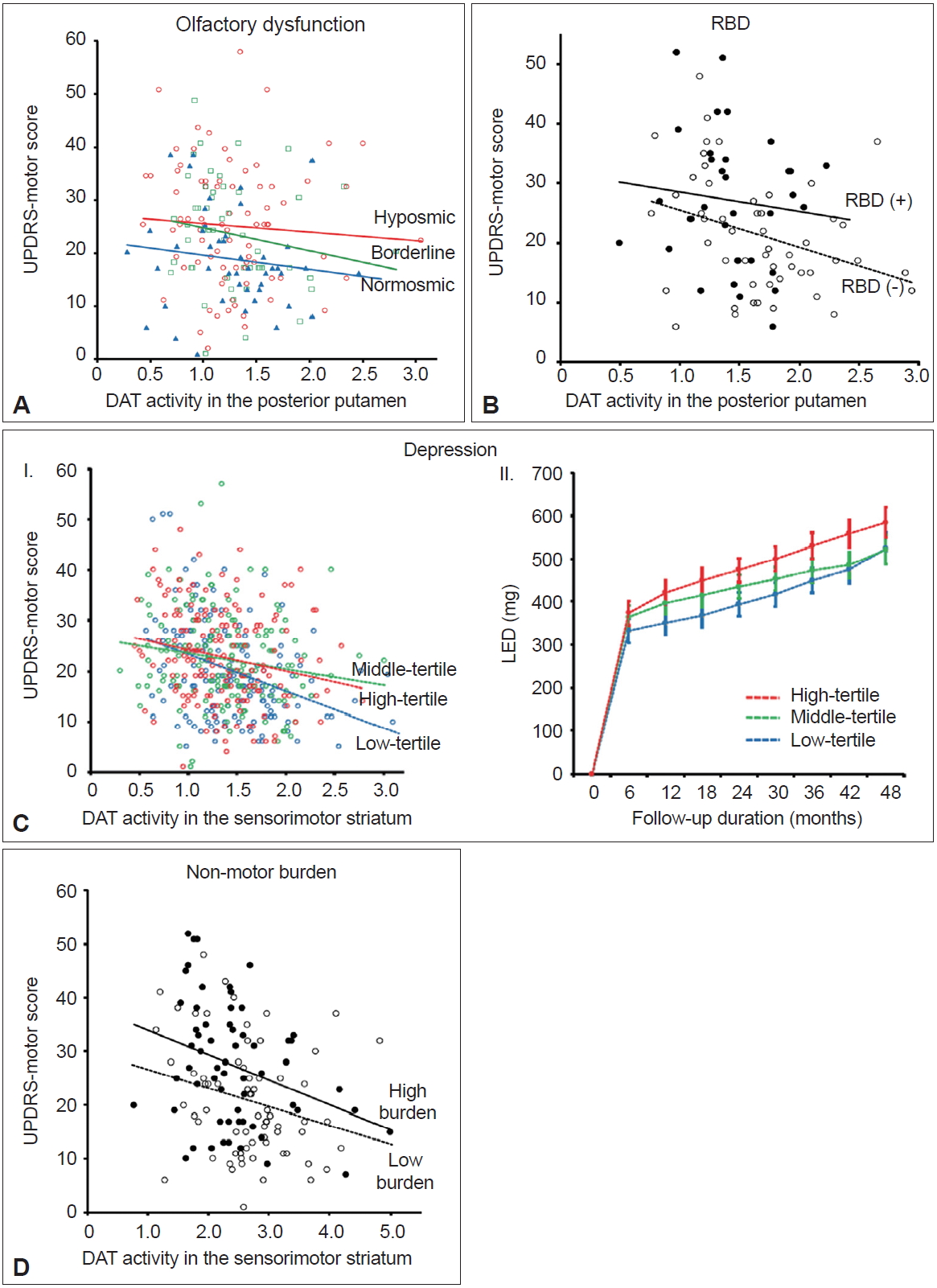
- Olfactory dysfunction is present in approximately 70–90% of patients with early-stage PD and can precede the onset of motor symptoms by several years [56-58]. The pathologic process of PD spreads from the olfactory bulb, anterior olfactory nucleus, and lower brainstem to different brain areas [59,60]. Olfactory dysfunction is typically nonprogressive once motor symptoms develop [57,61]; thus, predetermined olfactory involvement may have an impact on PD progression. Furthermore, the olfactory bulb is the main source for neurogenesis in adults [62]. Thus, patients with spared olfaction (i.e., normosmic PD) may have a greater potential for neurogenesis and greater MR against the neuropathological processes of PD compared to hyposmic PD. Among a total of 208 patients who performed a Cross Cultural Smell Identification Test (CCSIT), normosmic patients (CCSIT score ≥ 9; n = 53) exhibited fewer motor deficits after controlling for potential confounding factors, including DAT activity in the posterior putamen, compared to hyposmic patients (CCSIT score ≤ 6, n = 96) (Figure 4A) [8]. The LED during follow-up tended to be lower in normosmic compared with hyposmic PD patients. These findings suggest that normosmic PD is a unique clinical phenotype with greater MR and a more benign course compared to hyposmic PD.
- REM sleep behavioral disorder
- Rapid eye movement sleep behavior disorder (RBD) is an important biomarker of prodromal PD [63]. Polysomnographyproven RBD exhibits a 130-fold increased likelihood ratio for PD [64]. Accordingly, striatal DAT activity is reduced in patients with idiopathic RBD compared to healthy controls [65]. PD patients without RBD exhibited a different pattern of striatal DAT activity compared to those with RBD [66]. Furthermore, a prospective cohort study demonstrated that the presence of RBD in patients with PD at baseline indicates a more rapid disease progression of PD [67]. Therefore, PD without RBD may represent a benign motor phenotype of PD similar to normosmic PD. Among a total of 122 patients who performed the RBD Screening Questionnaire (RBDSQ) [68] at baseline, patients with clinically probable RBD (RBDSQ score ≥ 7, n = 39; a cut-off of 6/7 was used to minimize the false positives for the presence of RBD in patients with PD [69]) exhibited greater parkinsonian motor deficits predominantly in the less-affected side and axial symptoms and required higher LEDs during the follow-up period compared to those without clinically probable RBD (RBDSQ score ≤ 4, n = 58) [70]. These differences in motor deficits remained significant after controlling for DAT activity in the putamen and other confounding variables (Figure 4B), suggesting that the presence of RBD at baseline may represent a distinct PD subtype with a malignant motor phenotype and low MR in individuals with PD.
- Depression
- Depression is a representative nonmotor symptom (NMS) that may precede the onset of parkinsonian motor symptoms [63]; the presence of depression in early-stage PD has been proposed to result from the pathological involvement of the monoaminergic brainstem nuclei [71]. Accordingly, early accompaniment of depression in PD may indicate widespread involvement of PD pathologies, which subsequently may limit compensatory ability and reduce MR in individuals with PD. A previous study that showed more physical impairments in depressed than nondepressed patients with PD [72] supports the above hypothesis. A total of 474 patients who performed the Beck Depression Inventory (BDI) at baseline were divided into tertiles based on their BDI score [73]. The highest tertile group (BDI score ≥ 15; n = 157) showed more severe motor deficits and a lower level of cognitive performance than the lowest tertile group (BDI score ≤ 7, n = 158). This difference in motor deficits remained significant after controlling for DAT activity in the posterior putamen and other confounding factors (Figure 4C–I). In addition, the highest tertile group received higher LEDs for symptom control during follow-up than the lowest tertile group after controlling for age, sex, and initial motor deficit severity (Figure 4C–II). These findings indicate that the presence of depression at baseline represents reduced MR in PD.
- Nonmotor burden
- A variety of NMS are frequently accompanied by PD, which occur across all stages of PD, including the prodromal stage [74,75]. The involvement of PD-related pathology in widespread brainstem and cortical regions could be responsible for various NMS [75-77]. Therefore, patients with greater nonmotor burden at baseline may have more widespread involvement of PD pathology and limited MR in PD compared to those with fewer nonmotor burden. A cluster analysis study has demonstrated that nonmotor dominant patients with PD exhibit a more rapid motor progression than either pure-motor or mixed motor/nonmotor PD patients [78], which supports the above hypothesis. A total of 151 patients who performed the Korean version of Non-Motor Symptom Scale (K-NMSS) [79] at baseline were classified into two groups: high nonmotor burden group (K-NMSS score ≥ 41, n = 71) and low nonmotor burden group (K-NMSS score < 41, n = 80) [80]. Patients in the high nonmotor burden group were older, had a longer disease duration, exhibited more severe parkinsonian motor deficits, and received higher doses of dopaminergic medications during follow-up than those in the low nonmotor burden group despite similar levels of striatal DAT activity. The difference in motor deficits remained significant after controlling for potential confounding factors, including DAT activity, in the sensorimotor striatum (Figure 4D). These results suggest that low nonmotor burden at baseline represents a greater MR in PD compared to high nonmotor burden at baseline.
PRECLINICAL AND CLINICAL PRESENTATION AND MR
- Dopamine depletion patterns
- Selective and asymmetric involvement of the posterior putamen is the principal pathological feature of the nigrostriatal pathway in patients with PD [81-85]; however, the mechanism underlying this regional vulnerability remains unclear. The spatial patterns of striatal dopaminergic denervation are maintained as the disease progresses [83,84] and vary extensively among individuals with PD. Therefore, the pattern of striatal dopamine depletion might represent a consistent and specific characteristic of each patient with PD and could provide information on the clinical profiles; few studies have demonstrated that residual dopamine in the associative/limbic and contralateral striatum can act as a compensatory mechanism for parkinsonian motor symptoms [86,87]. A total of 634 patients were divided into tertile groups according to their patterns of striatal dopamine depletion, i.e., 1) the degree of dopamine loss found in the other striatal subregions compared to the posterior putamen [intersubregional ratio (ISR)] and 2) the interhemispheric asymmetry of dopamine deficits in the posterior putamen [asymmetry index (AI)] (Figure 5A–I) [88]. The highest tertile group of patients with PD according to AI exhibited milder parkinsonian motor signs than the lowest tertile group despite their greater decrease in DAT activity in the more affected posterior putamen (Figure 5A–II). In addition, the highest tertile group according to either ISR or AI values received lower doses of dopaminergic medications for symptom control than the corresponding lowest tertile group during the follow-up period (> 2 years). These findings suggest that the baseline patterns of striatal dopamine depletion can act as a marker for MR in PD, while high ISR and AI values represent a greater MR.
- White matter hyperintensity signals
- White matter hyperintensities (WMHs) are commonly observed in brain imaging studies of the healthy elderly [89]. Ample evidence has suggested that WMHs have a clinical impact on motor disability in the elderly [90], which might be associated with interruption of frontal subcortical motor circuits [91]. Moreover, white matter integrity appears to be related to the nigrostriatal synaptic dopamine function via common biological mechanisms [92]. Accordingly, WMH might contribute to the clinical severity in patients with PD. Indeed, a number of studies have demonstrated that severe WMH is associated with greater motor deficits, especially axial motor impairments in patients with PD [93-98], whereas some studies failed to demonstrate this association [99]. Our data revealed that the PD group with moderate to severe WMH (n = 109) exhibited more severe motor deficits than the PD group with minimal WMH (n = 227) despite comparable striatal DAT activity [43]. Furthermore, the PD group with moderate to severe WMH required higher doses of dopaminergic medications for symptom control compared to the PD group with minimal WMH (Figure 5B–I). The moderate to severe WMH group also exhibited an increased risk of developing freezing of gait (Figure 5B–II) [100]. These results suggest that baseline WMH severity can be used as an imaging marker for MR as well as a prognostic marker for motor outcomes in individuals with PD.
- Functional brain network associated with MR
- In patients with AD, several neuroimaging studies have demonstrated that neural substrates of CR are closely linked to AD pathology-prone regions [101-106]. We hypothesized that the neural correlates of MR in PD populations might be coupled with the network associated with motor function. Among 134 patients who performed resting state functional MRI at baseline, we calculated the ‘MR estimate’ of each patient based on the UPDRS motor scores and DAT activity in the posterior putamen using a residual model with high MR estimates indicating high MR [22]. Then, we applied a network-based statistic (NBS) analysis to identify the functional brain network associated with the MR estimate (i.e., MR network) using resting-state functional MRI data. NBS analysis identified that the MR network comprised of the basal ganglia (putamen, caudate, pallidum), inferior frontal cortex, insula, cerebellar vermis, hippocampus, and amygdala (Figure 5C–I), which could share the core components of the network associated with motor function in PD [107-110]. Patients with an increased degree of functional connectivity within the MR network exhibited a greater MR. Moreover, higher MR network strength (i.e., increased functional connectivity within the MR network) was associated with a slower longitudinal increase in doses of dopaminergic medications (Figure 5C–II). These findings suggest that functional connectivity within the MR network could indicate an individual’s capacity to cope with neurodegenerative processes in PD.
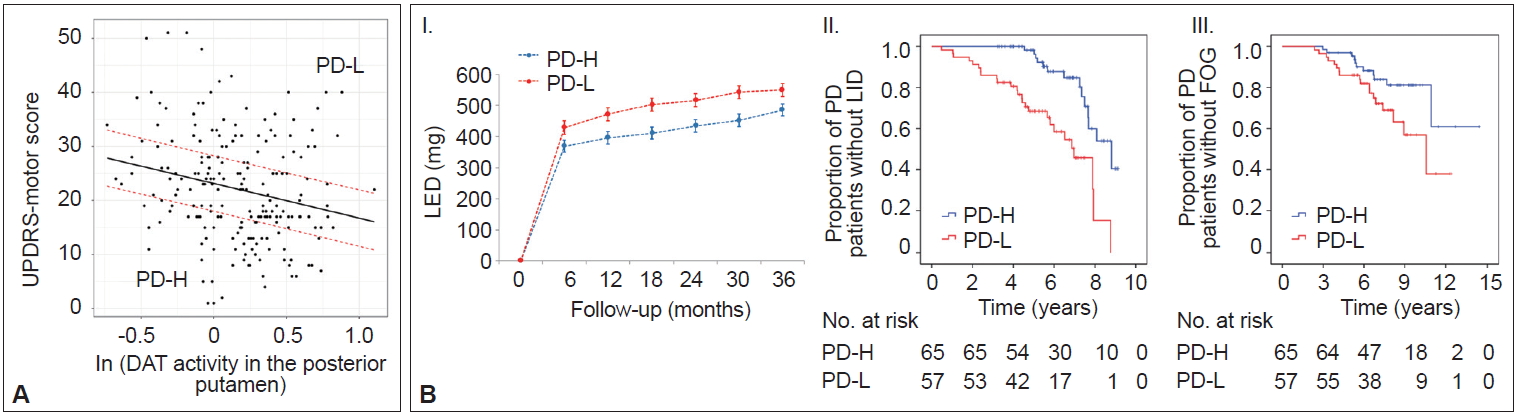
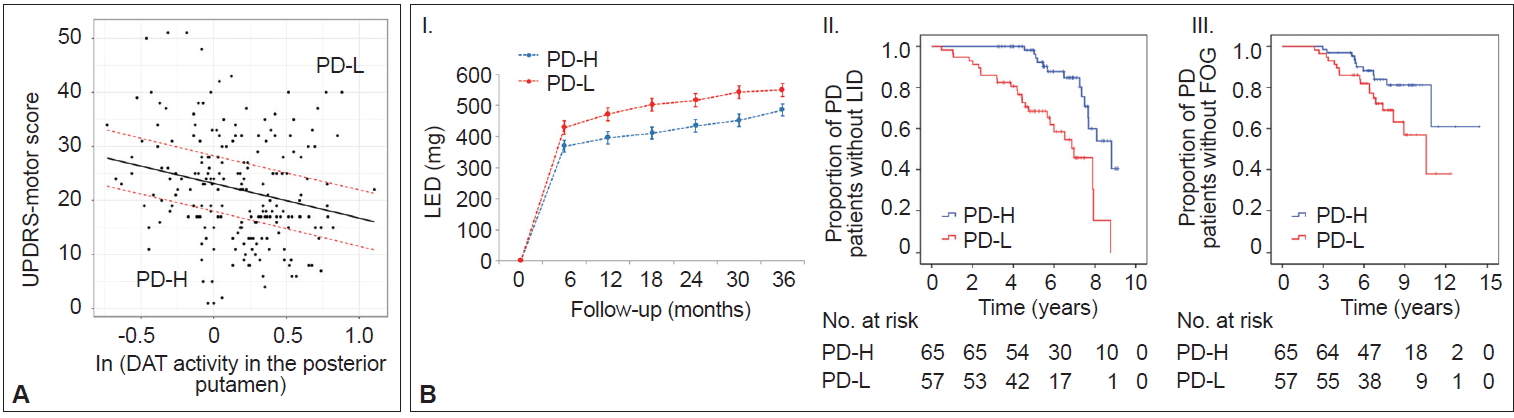
- Prognostic implications of MR
- In AD populations, it is well established that patients with high CR exhibit a greater capacity to tolerate AD pathology but exhibit a more rapid cognitive decline once the critical threshold is reached. This is presumably due to more advanced pathologies at the critical threshold coupled with the shorter time frame required to reach the point when AD pathology overwhelms cognitive function [2]. However, the impact of the initial MR on the long-term prognosis in PD remains unclear. A total of 205 patients were classified into two groups based on their MR estimate, including those with high MR (n = 65) and those with low MR (n = 57), which was determined by initial motor deficits and striatal DAT activity (Figure 6A) [111]. As expected, the low MR group exhibited higher baseline motor deficits than the high MR group despite having comparable levels of DAT activity in the posterior putamen. During the follow-up period, the low MR group received higher doses of dopaminergic medications for symptom control than the high MR group (Figure 6B–I). Moreover, the low MR group exhibited an increased risk of developing levodopa-induced dyskinesia and freezing of gait compared with the high MR group, which are important clinical milestones of disease progression in PD (Figure 6B–II and III). These results suggest that initial MR (i.e., the individual’s capacity to tolerate PD pathology) can be maintained with disease progression. Our observations are consistent with a passive reserve model (i.e., a greater capacity of preexisting neural substrates) rather than an active reserve model (i.e., more efficient neural compensations that might result in more rapid progression once parkinsonian symptoms manifest) in PD [21,22]. Therefore, factors enhancing MR (e.g., education [18] and physical activity [13]) may be protective against PD pathology throughout the disease course and can be used as preventive and therapeutic strategies against PD [112,113].
- Summary
- In this review, we discussed several factors that can enhance or reduce the MR in patients with PD (Table 1). In particular, some premorbid experiences (i.e., physical activity and education) and modifiable factors, such as BMI and WMH (which might be related to vascular risk factors), could modulate an individual’s capacity to tolerate PD pathology, which can be maintained throughout disease progression. Therefore, modification of these MR-related factors may be a reasonable treatment strategy for delaying parkinsonian symptom onset or improving the long-term motor outcomes of PD. Further studies are also needed to determine whether an active or passive reserve model will work in the strategy of enhancing each MR proxy.
NEUROIMAGING MARKERS AND MR
-
Conflicts of Interest
The authors have no financial conflicts of interest.
-
Ethical Standard
All procedures performed in our previous studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1975 Helsinki declaration and its later amendments or comparable ethical standards.
-
Author Contributions
Conceptualization: Young H. Sohn. Data curation: Seok Jong Chung, Jae Jung Lee, Phil Hyu Lee, Young H. Sohn. Formal analysis: Seok Jong Chung, Jae Jung Lee, Young H. Sohn. Investigation: all authors. Methodology: Seok Jong Chung, Young H. Sohn. Project administration: Young H. Sohn. Resources: Phil Hyu Lee, Young H. Sohn. Supervision: Young H. Sohn. Validation: Young H. Sohn. Visualization: Seok Jong Chung, Jae Jung Lee, Young H. Sohn. Writing—original draft: Seok Jong Chung, Young H. Sohn. Writing—review & editing: Jae Jung Lee, Phil Hyu Lee. Approval of final manuscript: all authors.
Notes
- We thank Mun Kyung Sunwoo, Jee Hyun Ham, Yoonju Lee, Dong Hyun Lee, and Su Jin Chung for contributions to a series of our previous works.
Acknowledgments

- 1. Pettigrew C, Soldan A. Defining cognitive reserve and implications for cognitive aging. Curr Neurol Neurosci Rep 2019;19:1.ArticlePubMedPMCPDF
- 2. Stern Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol 2012;11:1006–1012.ArticlePubMedPMC
- 3. Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 1991;114(Pt 5):2283–2301.ArticlePubMedPDF
- 4. Wilson RS, Nag S, Boyle PA, Hizel LP, Yu L, Buchman AS, et al. Neural reserve, neuronal density in the locus ceruleus, and cognitive decline. Neurology 2013;80:1202–1208.ArticlePubMedPMC
- 5. Palmer SJ, Ng B, Abugharbieh R, Eigenraam L, McKeown MJ. Motor reserve and novel area recruitment: amplitude and spatial characteristics of compensation in Parkinson’s disease. Eur J Neurosci 2009;29:2187–2196.ArticlePubMed
- 6. Gibb WR, Lees AJ. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry 1988;51:745–752.ArticlePubMedPMC
- 7. Oh M, Kim JS, Kim JY, Shin KH, Park SH, Kim HO, et al. Subregional patterns of preferential striatal dopamine transporter loss differ in Parkinson disease, progressive supranuclear palsy, and multiple-system atrophy. J Nucl Med 2012;53:399–406.ArticlePubMed
- 8. Lee DH, Oh JS, Ham JH, Lee JJ, Lee I, Lee PH, et al. Is normosmic Parkinson disease a unique clinical phenotype? Neurology 2015;85:1270–1275.ArticlePubMed
- 9. Smith AD, Zigmond MJ. Can the brain be protected through exercise? Lessons from an animal model of parkinsonism. Exp Neurol 2003;184:31–39.ArticlePubMed
- 10. Petzinger GM, Walsh JP, Akopian G, Hogg E, Abernathy A, Arevalo P, et al. Effects of treadmill exercise on dopaminergic transmission in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned mouse model of basal ganglia injury. J Neurosci 2007;27:5291–5300.ArticlePubMedPMC
- 11. Xu Q, Park Y, Huang X, Hollenbeck A, Blair A, Schatzkin A, et al. Physical activities and future risk of Parkinson disease. Neurology 2010;75:341–348.ArticlePubMedPMC
- 12. Chen H, Zhang SM, Schwarzschild MA, Hernán MA, Ascherio A. Physical activity and the risk of Parkinson disease. Neurology 2005;64:664–669.ArticlePubMed
- 13. Sunwoo MK, Lee JE, Hong JY, Ye BS, Lee HS, Oh JS, et al. Premorbid exercise engagement and motor reserve in Parkinson’s disease. Parkinsonism Relat Disord 2017;34:49–53.ArticlePubMed
- 14. Olsson TT, Svensson M, Hållmarker U, James S, Deierborg T. Delayed clinical manifestation of Parkinson’s disease among physically active: do participants in a long-distance ski race have a motor reserve? J Parkinsons Dis 2020;10:267–274.ArticlePubMedPMC
- 15. Taylor CA, Saint-Hilaire MH, Cupples LA, Thomas CA, Burchard AE, Feldman RG, et al. Environmental, medical, and family history risk factors for Parkinson’s disease: a New England-based case control study. Am J Med Genet 1999;88:742–749.ArticlePubMed
- 16. Rocca WA, Anderson DW, Meneghini F, Grigoletto F, Morgante L, Reggio A, et al. Occupation, education, and Parkinson’s disease: a case-control study in an Italian population. Mov Disord 1996;11:201–206.ArticlePubMed
- 17. Frigerio R, Elbaz A, Sanft KR, Peterson BJ, Bower JH, Ahlskog JE, et al. Education and occupations preceding Parkinson disease: a population-based case-control study. Neurology 2005;65:1575–1583.ArticlePubMed
- 18. Sunwoo MK, Hong JY, Lee JJ, Lee PH, Sohn YH. Does education modify motor compensation in Parkinson’s disease? J Neurol Sci 2016;362:118–120.ArticlePubMed
- 19. Kotagal V, Bohnen NI, Müller ML, Koeppe RA, Frey KA, Langa KM, et al. Educational attainment and motor burden in Parkinson’s disease. Mov Disord 2015;30:1143–1147.ArticlePubMedPMC
- 20. Blume J, Rothenfusser E, Schlaier J, Bogdahn U, Lange M. Educational attainment and motor burden in advanced Parkinson’s disease—The emerging role of education in motor reserve. J Neurol Sci 2017;381:141–143.ArticlePubMed
- 21. Lee PC, Artaud F, Cormier-Dequaire F, Rascol O, Durif F, Derkinderen P, et al. Examining the reserve hypothesis in Parkinson’s disease: a longitudinal study. Mov Disord 2019;34:1663–1671.ArticlePubMed
- 22. Chung SJ, Kim HR, Jung JH, Lee PH, Jeong Y, Sohn YH. Identifying the functional brain network of motor reserve in early Parkinson’s disease. Mov Disord 2020;35:577–586.ArticlePubMed
- 23. Allam MF, Campbell MJ, Hofman A, Del Castillo AS, Fernández-Crehuet Navajas R. Smoking and Parkinson’s disease: systematic review of prospective studies. Mov Disord 2004;19:614–621.ArticlePubMed
- 24. Allam MF, Campbell MJ, Del Castillo AS, Fernández-Crehuet Navajas R. Parkinson’s disease protects against smoking? Behav Neurol 2004;15:65–71.ArticlePubMedPDF
- 25. Gorell JM, Rybicki BA, Johnson CC, Peterson EL. Smoking and Parkinson’s disease: a dose-response relationship. Neurology 1999;52:115–119.ArticlePubMed
- 26. Shahi GS, Das NP, Moochhala SM. 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity: partial protection against striatonigral dopamine depletion in C57BL/6J mice by cigarette smoke exposure and by beta-naphthoflavone-pretreatment. Neurosci Lett 1991;127:247–250.ArticlePubMed
- 27. Carr LA, Rowell PP. Attenuation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity by tobacco smoke. Neuropharmacology 1990;29:311–314.ArticlePubMed
- 28. Lee Y, Oh JS, Chung SJ, Chung SJ, Kim SJ, Nam CM, et al. Does smoking impact dopamine neuronal loss in de novo Parkinson disease? Ann Neurol 2017;82:850–854.ArticlePubMed
- 29. van Rooden SM, Heiser WJ, Kok JN, Verbaan D, van Hilten JJ, Marinus J. The identification of Parkinson’s disease subtypes using cluster analysis: a systematic review. Mov Disord 2010;25:969–978.ArticlePubMed
- 30. Kempster PA, O’Sullivan SS, Holton JL, Revesz T, Lees AJ. Relationships between age and late progression of Parkinson’s disease: a clinico-pathological study. Brain 2010;133:1755–1762.ArticlePubMedPDF
- 31. Wickremaratchi MM, Ben-Shlomo Y, Morris HR. The effect of onset age on the clinical features of Parkinson’s disease. Eur J Neurol 2009;16:450–456.ArticlePubMed
- 32. Liu SY, Wu JJ, Zhao J, Huang SF, Wang YX, Ge JJ, et al. Onset-related subtypes of Parkinson’s disease differ in the patterns of striatal dopaminergic dysfunction: a positron emission tomography study. Parkinsonism Relat Disord 2015;21:1448–1453.ArticlePubMed
- 33. Shih MC, Franco de Andrade LA, Amaro E Jr, Felicio AC, Ferraz HB, Wagner J, et al. Higher nigrostriatal dopamine neuron loss in early than late onset Parkinson’s disease?—A [99mTc]-TRODAT-1 SPECT study. Mov Disord 2007;22:863–866.ArticlePubMed
- 34. de la Fuente-Fernández R, Schulzer M, Kuramoto L, Cragg J, Ramachandiran N, Au WL, et al. Age-specific progression of nigrostriatal dysfunction in Parkinson’s disease. Ann Neurol 2011;69:803–810.ArticlePubMed
- 35. Pagano G, Ferrara N, Brooks DJ, Pavese N. Age at onset and Parkinson disease phenotype. Neurology 2016;86:1400–1407.ArticlePubMedPMC
- 36. Chung SJ, Yoo HS, Lee YH, Lee PH, Sohn YH. Heterogeneous patterns of striatal dopamine loss in patients with young- versus old-onset Parkinson’s disease: impact on clinical features. J Mov Disord 2019;12:113–119.ArticlePubMedPMCPDF
- 37. Wooten GF, Currie LJ, Bovbjerg VE, Lee JK, Patrie J. Are men at greater risk for Parkinson’s disease than women? J Neurol Neurosurg Psychiatry 2004;75:637–639.ArticlePubMedPMC
- 38. Mayeux R, Marder K, Cote LJ, Denaro J, Hemenegildo N, Mejia H, et al. The frequency of idiopathic Parkinson’s disease by age, ethnic group, and sex in northern Manhattan, 1988-1993. Am J Epidemiol 1995;142:820–827.ArticlePubMedPDF
- 39. Haaxma CA, Bloem BR, Borm GF, Oyen WJ, Leenders KL, Eshuis S, et al. Gender differences in Parkinson’s disease. J Neurol Neurosurg Psychiatry 2007;78:819–824.ArticlePubMed
- 40. Growdon JH, Kieburtz K, McDermott MP, Panisset M, Friedman JH. Levodopa improves motor function without impairing cognition in mild non-demented Parkinson’s disease patients. Parkinson study group. Neurology 1998;50:1327–1331.ArticlePubMed
- 41. Lyons KE, Hubble JP, Tröster AI, Pahwa R, Koller WC. Gender differences in Parkinson’s disease. Clin Neuropharmacol 1998;21:118–121.PubMed
- 42. Chung SJ, Yoo HS, Lee HS, Jeong HE, Kim SJ, Oh JS, et al. Does late levodopa administration delay the development of dyskinesia in patients with de novo Parkinson’s disease? CNS Drugs 2018;32:971–979.ArticlePubMedPDF
- 43. Chung SJ, Yoo HS, Lee YH, Jung JH, Baik K, Ye BS, et al. White matter hyperintensities and risk of levodopa-induced dyskinesia in Parkinson’s disease. Ann Clin Transl Neurol 2020;7:229–238.ArticlePubMedPMC
- 44. Kim HJ, Mason S, Foltynie T, Winder-Rhodes S, Barker RA, Williams-Gray CH. Motor complications in Parkinson’s disease: 13-year follow-up of the CamPaIGN cohort. Mov Disord 2020;35:185–190.ArticlePubMed
- 45. Lee JJ, Ham JH, Lee PH, Sohn YH. Gender differences in age-related striatal dopamine depletion in Parkinson’s disease. J Mov Disord 2015;8:130–135.ArticlePubMedPMCPDF
- 46. Triggs WJ, Calvanio R, Levine M, Heaton RK, Heilman KM. Predicting hand preference with performance on motor tasks. Cortex 2000;36:679–689.ArticlePubMed
- 47. Hammond G. Correlates of human handedness in primary motor cortex: a review and hypothesis. Neurosci Biobehav Rev 2002;26:285–292.ArticlePubMed
- 48. Nudo RJ, Jenkins WM, Merzenich MM, Prejean T, Grenda R. Neurophysiological correlates of hand preference in primary motor cortex of adult squirrel monkeys. J Neurosci 1992;12:2918–2947.ArticlePubMedPMC
- 49. Volkmann J, Schnitzler A, Witte OW, Freund H. Handedness and asymmetry of hand representation in human motor cortex. J Neurophysiol 1998;79:2149–2154.ArticlePubMed
- 50. Hoehn MM, Yahr MD. Parkinsonism: onset, progression and mortality. Neurology 1967;17:427–442.ArticlePubMed
- 51. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–184.ArticlePubMedPMC
- 52. Ham JH, Lee JJ, Kim JS, Lee PH, Sohn YH. Is dominant-side onset associated with a better motor compensation in Parkinson’s disease? Mov Disord 2015;30:1921–1925.ArticlePubMed
- 53. Chen H, Zhang SM, Hernán MA, Willett WC, Ascherio A. Weight loss in Parkinson’s disease. Ann Neurol 2003;53:676–679.ArticlePubMed
- 54. van der Marck MA, Dicke HC, Uc EY, Kentin ZH, Borm GF, Bloem BR, et al. Body mass index in Parkinson’s disease: a meta-analysis. Parkinsonism Relat Disord 2012;18:263–267.ArticlePubMed
- 55. Lee JJ, Oh JS, Ham JH, Lee DH, Lee I, Sohn YH, et al. Association of body mass index and the depletion of nigrostriatal dopamine in Parkinson’s disease. Neurobiol Aging 2016;38:197–204.ArticlePubMed
- 56. Doty RL. Olfactory dysfunction in Parkinson disease. Nat Rev Neurol 2012;8:329–339.ArticlePubMedPDF
- 57. Doty RL, Deems DA, Stellar S. Olfactory dysfunction in parkinsonism: a general deficit unrelated to neurologic signs, disease stage, or disease duration. Neurology 1988;38:1237–1244.ArticlePubMed
- 58. Hawkes CH, Shephard BC, Daniel SE. Olfactory dysfunction in Parkinson’s disease. J Neurol Neurosurg Psychiatry 1997;62:436–446.ArticlePubMedPMC
- 59. Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003;24:197–211.ArticlePubMed
- 60. Hawkes CH. Parkinson’s disease and aging: same or different process? Mov Disord 2008;23:47–53.ArticlePubMed
- 61. Doty RL, Stern MB, Pfeiffer C, Gollomp SM, Hurtig HI. Bilateral olfactory dysfunction in early stage treated and untreated idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry 1992;55:138–142.ArticlePubMedPMC
- 62. Marxreiter F, Regensburger M, Winkler J. Adult neurogenesis in Parkinson’s disease. Cell Mol Life Sci 2013;70:459–473.ArticlePubMedPDF
- 63. Postuma RB, Aarsland D, Barone P, Burn DJ, Hawkes CH, Oertel W, et al. Identifying prodromal Parkinson’s disease: pre-motor disorders in Parkinson’s disease. Mov Disord 2012;27:617–626.ArticlePubMed
- 64. Berg D, Postuma RB, Adler CH, Bloem BR, Chan P, Dubois B, et al. MDS research criteria for prodromal Parkinson’s disease. Mov Disord 2015;30:1600–1611.ArticlePubMed
- 65. Eisensehr I, Linke R, Noachtar S, Schwarz J, Gildehaus FJ, Tatsch K. Reduced striatal dopamine transporters in idiopathic rapid eye movement sleep behaviour disorder. Comparison with Parkinson’s disease and controls. Brain 2000;123(Pt 6):1155–1160.ArticlePubMedPDF
- 66. Arnaldi D, De Carli F, Picco A, Ferrara M, Accardo J, Bossert I, et al. Nigro-caudate dopaminergic deafferentation: a marker of REM sleep behavior disorder? Neurobiol Aging 2015;36:3300–3305.ArticlePubMed
- 67. Fereshtehnejad SM, Romenets SR, Anang JB, Latreille V, Gagnon JF, Postuma RB. New clinical subtypes of Parkinson disease and their longitudinal progression: a prospective cohort comparison with other phenotypes. JAMA Neurol 2015;72:863–873.ArticlePubMed
- 68. Stiasny-Kolster K, Mayer G, Schäfer S, Möller JC, Heinzel-Gutenbrunner M, Oertel WH. The REM sleep behavior disorder screening questionnaire—A new diagnostic instrument. Mov Disord 2007;22:2386–2393.ArticlePubMed
- 69. Chahine LM, Daley J, Horn S, Colcher A, Hurtig H, Cantor C, et al. Questionnaire-based diagnosis of REM sleep behavior disorder in Parkinson’s disease. Mov Disord 2013;28:1146–1149.ArticlePubMed
- 70. Chung SJ, Lee Y, Lee JJ, Lee PH, Sohn YH. Rapid eye movement sleep behaviour disorder and striatal dopamine depletion in patients with Parkinson’s disease. Eur J Neurol 2017;24:1314–1319.ArticlePubMed
- 71. Aarsland D, Påhlhagen S, Ballard CG, Ehrt U, Svenningsson P. Depression in Parkinson disease--epidemiology, mechanisms and management. Nat Rev Neurol 2011;8:35–47.ArticlePubMedPDF
- 72. Ravina B, Camicioli R, Como PG, Marsh L, Jankovic J, Weintraub D, et al. The impact of depressive symptoms in early Parkinson disease. Neurology 2007;69:342–347.ArticlePubMedPMC
- 73. Lee Y, Oh JS, Chung SJ, Lee JJ, Chung SJ, Moon H, et al. The presence of depression in de novo Parkinson’s disease reflects poor motor compensation. PLoS One 2018;13:e0203303. ArticlePubMedPMC
- 74. Chaudhuri KR, Healy DG, Schapira AH. Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol 2006;5:235–245.ArticlePubMed
- 75. Chaudhuri KR, Schapira AH. Non-motor symptoms of Parkinson’s disease: dopaminergic pathophysiology and treatment. Lancet Neurol 2009;8:464–474.ArticlePubMed
- 76. Wolters ECh, Braak H. Parkinson’s disease: premotor clinico-pathological correlations. J Neural Transm Suppl 2006;70:309–319.Article
- 77. Kingsbury AE, Bandopadhyay R, Silveira-Moriyama L, Ayling H, Kallis C, Sterlacci W, et al. Brain stem pathology in Parkinson’s disease: an evaluation of the Braak staging model. Mov Disord 2010;25:2508–2515.ArticlePubMed
- 78. Erro R, Vitale C, Amboni M, Picillo M, Moccia M, Longo K, et al. The heterogeneity of early Parkinson’s disease: a cluster analysis on newly diagnosed untreated patients. PLoS One 2013;8:e70244. ArticlePubMedPMC
- 79. Koh SB, Kim JW, Ma HI, Ahn TB, Cho JW, Lee PH, et al. Validation of the Korean-version of the nonmotor symptoms scale for Parkinson’s disease. J Clin Neurol 2012;8:276–283.ArticlePubMedPMC
- 80. Chung SJ, Lee JJ, Ham JH, Ye BS, Lee PH, Sohn YH. Striatal dopamine depletion patterns and early non-motor burden in Parkinsons disease. PLoS One 2016;11:e0161316. ArticlePubMedPMC
- 81. Brooks DJ, Ibanez V, Sawle GV, Quinn N, Lees AJ, Mathias CJ, et al. Differing patterns of striatal 18F-dopa uptake in Parkinson’s disease, multiple system atrophy, and progressive supranuclear palsy. Ann Neurol 1990;28:547–555.ArticlePubMed
- 82. Knable MB, Jones DW, Coppola R, Hyde TM, Lee KS, Gorey J, et al. Lateralized differences in iodine-123-IBZM uptake in the basal ganglia in asymmetric Parkinson’s disease. J Nucl Med 1995;36:1216–1225.PubMed
- 83. Nandhagopal R, Kuramoto L, Schulzer M, Mak E, Cragg J, Lee CS, et al. Longitudinal progression of sporadic Parkinson’s disease: a multitracer positron emission tomography study. Brain 2009;132:2970–2979.ArticlePubMedPDF
- 84. Djaldetti R, Lorberboym M, Karmon Y, Treves TA, Ziv I, Melamed E. Residual striatal dopaminergic nerve terminals in very long-standing Parkinson’s disease: a single photon emission computed tomography imaging study. Mov Disord 2011;26:327–330.ArticlePubMed
- 85. Scherfler C, Seppi K, Mair KJ, Donnemiller E, Virgolini I, Wenning GK, et al. Left hemispheric predominance of nigrostriatal dysfunction in Parkinson’s disease. Brain 2012;135:3348–3354.ArticlePubMedPDF
- 86. Blesa J, Juri C, García-Cabezas MÁ, Adánez R, Sánchez-González MÁ, Cavada C, et al. Inter-hemispheric asymmetry of nigrostriatal dopaminergic lesion: a possible compensatory mechanism in Parkinson’s disease. Front Syst Neurosci 2011;5:92.ArticlePubMedPMC
- 87. Schneider JS, Rothblat DS, DiStefano L. Volume transmission of dopamine over large distances may contribute to recovery from experimental parkinsonism. Brain Res 1994;643:86–91.ArticlePubMed
- 88. Chung SJ, Yoo HS, Lee HS, Oh JS, Kim JS, Sohn YH, et al. The pattern of striatal dopamine depletion as a prognostic marker in de novo Parkinson disease. Clin Nucl Med 2018;43:787–792.ArticlePubMed
- 89. de Leeuw FE, de Groot JC, Achten E, Oudkerk M, Ramos LM, Heijboer R, et al. Prevalence of cerebral white matter lesions in elderly people: a population based magnetic resonance imaging study. The Rotterdam scan study. J Neurol Neurosurg Psychiatry 2001;70:9–14.ArticlePubMedPMC
- 90. van der Holst HM, van Uden IW, Tuladhar AM, de Laat KF, van Norden AG, Norris DG, et al. Cerebral small vessel disease and incident parkinsonism: the RUN DMC study. Neurology 2015;85:1569–1577.ArticlePubMedPMC
- 91. Blahak C, Baezner H, Pantoni L, Poggesi A, Chabriat H, Erkinjuntti T, et al. Deep frontal and periventricular age related white matter changes but not basal ganglia and infratentorial hyperintensities are associated with falls: cross sectional results from the LADIS study. J Neurol Neurosurg Psychiatry 2009;80:608–613.ArticlePubMed
- 92. Rodriguez-Perez AI, Dominguez-Meijide A, Lanciego JL, Guerra MJ, Labandeira-Garcia JL. Dopaminergic degeneration is enhanced by chronic brain hypoperfusion and inhibited by angiotensin receptor blockage. Age (Dordr) 2013;35:1675–1690.ArticlePubMedPDF
- 93. Piccini P, Pavese N, Canapicchi R, Paoli C, Del Dotto P, Puglioli M, et al. White matter hyperintensities in Parkinson’s disease. Clinical correlations. Arch Neurol 1995;52:191–194.ArticlePubMed
- 94. Sohn YH, Kim JS. The influence of white matter hyperintensities on the clinical features of Parkinson’s disease. Yonsei Med J 1998;39:50–55.ArticlePubMed
- 95. Lee SJ, Kim JS, Lee KS, An JY, Kim W, Kim YI, et al. The severity of leukoaraiosis correlates with the clinical phenotype of Parkinson’s disease. Arch Gerontol Geriatr 2009;49:255–259.ArticlePubMed
- 96. Bohnen NI, Müller ML, Zarzhevsky N, Koeppe RA, Bogan CW, Kilbourn MR, et al. Leucoaraiosis, nigrostriatal denervation and motor symptoms in Parkinson’s disease. Brain 2011;134:2358–2365.ArticlePubMedPMCPDF
- 97. Kotagal V, Albin RL, Müller ML, Koeppe RA, Frey KA, Bohnen NI. Modifiable cardiovascular risk factors and axial motor impairments in Parkinson disease. Neurology 2014;82:1514–1520.ArticlePubMedPMC
- 98. Arena JE, Cerquetti D, Rossi M, Chaves H, Rollan C, Dossi DE, et al. Influence of white matter MRI hyper-intensities on acute l-dopa response in patients with Parkinson’s disease. Parkinsonism Relat Disord 2016;24:126–128.ArticlePubMed
- 99. Herman T, Rosenberg-Katz K, Jacob Y, Auriel E, Gurevich T, Giladi N, et al. White matter hyperintensities in Parkinson’s disease: do they explain the disparity between the postural instability gait difficulty and tremor dominant subtypes? PLoS One 2013;8:e55193. ArticlePubMedPMC
- 100. Chung SJ, Lee YH, Yoo HS, Oh JS, Kim JS, Ye BS, et al. White matter hyperintensities as a predictor of freezing of gait in Parkinson’s disease. Parkinsonism Relat Disord 2019;66:105–109.ArticlePubMed
- 101. Kemppainen NM, Aalto S, Karrasch M, Någren K, Savisto N, Oikonen V, et al. Cognitive reserve hypothesis: Pittsburgh compound B and fluorodeoxyglucose positron emission tomography in relation to education in mild Alzheimer’s disease. Ann Neurol 2008;63:112–118.ArticlePubMed
- 102. Liu Y, Julkunen V, Paajanen T, Westman E, Wahlund LO, Aitken A, et al. Education increases reserve against Alzheimer’s disease--evidence from structural MRI analysis. Neuroradiology 2012;54:929–938.ArticlePubMedPMC
- 103. Ewers M, Insel PS, Stern Y, Weiner MW; Alzheimer’s Disease Neuroimaging Initiative (ADNI). Cognitive reserve associated with FDG-PET in preclinical Alzheimer disease. Neurology 2013;80:1194–1201.ArticlePubMedPMC
- 104. Garibotto V, Tettamanti M, Marcone A, Florea I, Panzacchi A, Moresco R, et al. Cholinergic activity correlates with reserve proxies in Alzheimer’s disease. Neurobiol Aging 2013;34:2694.e13–e18.ArticlePubMed
- 105. Hoenig MC, Bischof GN, Hammes J, Faber J, Fliessbach K, van Eimeren T, et al. Tau pathology and cognitive reserve in Alzheimer’s disease. Neurobiol Aging 2017;57:1–7.ArticlePubMed
- 106. Lee DH, Lee P, Seo SW, Roh JH, Oh M, Oh JS, et al. Neural substrates of cognitive reserve in Alzheimer’s disease spectrum and normal aging. Neuroimage 2019;186:690–702.ArticlePubMed
- 107. Groenewegen HJ. The basal ganglia and motor control. Neural Plast 2003;10:107–120.ArticlePubMedPMCPDF
- 108. Wu T, Hallett M. The cerebellum in Parkinson’s disease. Brain 2013;136:696–709.ArticlePubMedPMCPDF
- 109. Swann NC, Cai W, Conner CR, Pieters TA, Claffey MP, George JS, et al. Roles for the pre-supplementary motor area and the right inferior frontal gyrus in stopping action: electrophysiological responses and functional and structural connectivity. Neuroimage 2012;59:2860–2870.ArticlePubMed
- 110. Chikama M, McFarland NR, Amaral DG, Haber SN. Insular cortical projections to functional regions of the striatum correlate with cortical cytoarchitectonic organization in the primate. J Neurosci 1997;17:9686–9705.ArticlePubMedPMC
- 111. Chung SJ, Yoo HS, Lee YH, Lee HS, Lee PH, Sohn YH. Initial motor reserve and long-term prognosis in Parkinson’s disease. Neurobiol Aging 2020;92:1–6.ArticlePubMed
- 112. Xu W, Yu JT, Tan MS, Tan L. Cognitive reserve and Alzheimer’s disease. Mol Neurobiol 2015;51:187–208.ArticlePubMedPDF
- 113. Paillard T, Rolland Y, de Souto Barreto P. Protective effects of physical exercise in Alzheimer’s disease and Parkinson’s disease: a narrative review. J Clin Neurol 2015;11:212–219.ArticlePubMedPMC
REFERENCES
Figure & Data
References
Citations
- How long have you exercised in your life? The effect of motor reserve and current physical activity on cognitive performance
Veronica Pucci, Carolina Guerra, Amanda Barsi, Massimo Nucci, Sara Mondini
Journal of the International Neuropsychological Society.2024; 30(1): 11. CrossRef - Hippocampal Perfusion Affects Motor and Cognitive Functions in Parkinson Disease: An Early Phase 18F‐FP‐CIT Positron Emission Tomography Study
Min Young Chun, Seok Jong Chung, Su Hong Kim, Chan Wook Park, Seong Ho Jeong, Hye Sun Lee, Phil Hyu Lee, Young H. Sohn, Yong Jeong, Yun Joong Kim
Annals of Neurology.2024; 95(2): 388. CrossRef - Clinical severity in Parkinson’s disease is determined by decline in cortical compensation
Martin E Johansson, Ivan Toni, Roy P C Kessels, Bastiaan R Bloem, Rick C Helmich
Brain.2024; 147(3): 871. CrossRef - Differences in [123I]Ioflupane Striatal Binding Between African American and White Patients
Juebin Huang, Kevin J. Sullivan, Vani Vijayakumar
Journal of Nuclear Medicine Technology.2024; : jnmt.123.265806. CrossRef - Plasma extracellular vesicle synaptic proteins as biomarkers of clinical progression in patients with Parkinson’s disease
Chien-Tai Hong, Chen-Chih Chung, Ruan-Ching Yu, Lung Chan
eLife.2024;[Epub] CrossRef - Plasma extracellular vesicle synaptic proteins as biomarkers of clinical progression in patients with Parkinson’s disease
Chien-Tai Hong, Chen-Chih Chung, Ruan-Ching Yu, Lung Chan
eLife.2024;[Epub] CrossRef - Considering the response in addition to the challenge – a narrative review in appraisal of a motor reserve framework
Daniel Zeller, Shawn Hiew, Thorsten Odorfer, Carine Nguemeni
Aging.2024; 16(6): 5772. CrossRef - The greatest loss of unpleasant smells may be related to the risk of more severe PD symptoms
Shih-Chi Chiu, Ting-Chun Fang, Hsin-Bei Lei, Yu-Hsuan Lin, Ming-Hong Chang, Yi-Jen Guo
Frontiers in Neurology.2024;[Epub] CrossRef - Lifestyle Modulators of Neuroplasticity in Parkinson’s Disease: Evidence
in Human Neuroimaging Studies
Silvia Paola Caminiti, Silvia Gallo, Federico Menegon, Andrea Naldi, Cristoforo Comi, Giacomo Tondo
CNS & Neurological Disorders - Drug Targets.2024; 23(5): 602. CrossRef - Motor progression marker for newly diagnosed drug‐naïve patients with Parkinson's disease: A resting‐state functional MRI study
Yanbing Hou, Lingyu Zhang, Ruwei Ou, Qianqian Wei, Xiaojing Gu, Kuncheng Liu, Junyu Lin, Tianmi Yang, Yi Xiao, Qiyong Gong, Huifang Shang
Human Brain Mapping.2023; 44(3): 901. CrossRef - The Concept of Motor Reserve in Parkinson's Disease: New Wine in Old Bottles?
Merle C. Hoenig, Verena Dzialas, Alexander Drzezga, Thilo van Eimeren
Movement Disorders.2023; 38(1): 16. CrossRef - Patterns of striatal dopamine depletion and motor deficits in de novo Parkinson’s disease
Seong Ho Jeong, Chan Wook Park, Hye Sun Lee, Yun Joong Kim, Mijin Yun, Phil Hyu Lee, Young H. Sohn, Seok Jong Chung
Journal of Neural Transmission.2023; 130(1): 19. CrossRef - Sex Differences in Brain Structure in de novo Parkinson’s Disease: A Cross-Sectional and Longitudinal Neuroimaging Study
Hui Li, Xuejia Jia, Min Chen, Xiuqin Jia, Qi Yang
Journal of Parkinson's Disease.2023; 13(5): 785. CrossRef - Exploring the Complex Phenotypes of Impaired Finger Dexterity in Mild-to-moderate Stage Parkinson’s Disease: A Time-Series Analysis
Pattamon Panyakaew, Kotchakorn Duangjino, Apiwoot Kerddonfag, Teerit Ploensin, Krerk Piromsopa, Chanon Kongkamol, Roongroj Bhidayasiri
Journal of Parkinson's Disease.2023; 13(6): 975. CrossRef - Prevention of Falls in Parkinson's Disease: Guidelines and Gaps
Richard Camicioli, Meg E. Morris, Frederico Pieruccini‐Faria, Manuel Montero‐Odasso, Surim Son, David Buzaglo, Jeffrey M. Hausdorff, Alice Nieuwboer
Movement Disorders Clinical Practice.2023; 10(10): 1459. CrossRef - The incidence of deep venous thrombosis in Parkinson’s disease
Emine Afsin, Zeliha Coşgun, Ramazan Kurul, Şule Aydın Türkoğlu
Neurological Research.2023; 45(11): 1050. CrossRef - Premorbid Educational Attainment and Long-Term Motor Prognosis in Parkinson’s Disease
Seong Ho Jeong, Seok Jong Chung, Han Soo Yoo, Jin Ho Jung, Kyoungwon Baik, Yang Hyun Lee, Phil Hyu Lee, Young H. Sohn
Journal of Parkinson's Disease.2022; 12(1): 129. CrossRef - Parkinsonism and cerebrovascular disease
Manisha Narasimhan, Raymond Schwartz, Glenda Halliday
Journal of the Neurological Sciences.2022; 433: 120011. CrossRef - Impact of α‐synuclein spreading on the nigrostriatal dopaminergic pathway depends on the onset of the pathology
Fanfan Sun, Armando G. Salinas, Severin Filser, Sonja Blumenstock, Jose Medina‐Luque, Jochen Herms, Carmelo Sgobio
Brain Pathology.2022;[Epub] CrossRef - Premorbid cancer and motor reserve in patients with Parkinson’s disease
Yoon-Sang Oh, Sang-Won Yoo, Chul Hyoung Lyoo, Kwang-Soo Lee, Joong-Seok Kim
Scientific Reports.2022;[Epub] CrossRef - Behavioral Reserve in Behavioral Variant Frontotemporal Dementia
Su Hong Kim, Yae Ji Kim, Byung Hwa Lee, Peter Lee, Ji Hyung Park, Sang Won Seo, Yong Jeong
Frontiers in Aging Neuroscience.2022;[Epub] CrossRef - Identifying the white matter structural network of motor reserve in early Parkinson's disease
Yae Ji Kim, Chan Wook Park, Hye Won Shin, Hye Sun Lee, Yun Joong Kim, Mijin Yun, Phil Hyu Lee, Young H. Sohn, Yong Jeong, Seok Jong Chung
Parkinsonism & Related Disorders.2022; 102: 108. CrossRef - Comparison of disease progression between brain-predominant Parkinson's disease versus Parkinson's disease with body-involvement phenotypes
Dong-Woo Ryu, Sang-Won Yoo, Yoon-Sang Oh, Kwang-Soo Lee, Seunggyun Ha, Joong-Seok Kim
Neurobiology of Disease.2022; 174: 105883. CrossRef - Genetically-informed prediction of short-term Parkinson’s disease progression
Hossein J. Sadaei, Aldo Cordova-Palomera, Jonghun Lee, Jaya Padmanabhan, Shang-Fu Chen, Nathan E. Wineinger, Raquel Dias, Daria Prilutsky, Sandor Szalma, Ali Torkamani
npj Parkinson's Disease.2022;[Epub] CrossRef - Potential Link Between Cognition and Motor Reserve in Patients With Parkinson’s Disease
Seok Jong Chung, Yae Ji Kim, Yun Joong Kim, Hye Sun Lee, Mijin Yun, Phil Hyu Lee, Yong Jeong, Young H. Sohn
Journal of Movement Disorders.2022; 15(3): 249. CrossRef - Local striatal volume and motor reserve in drug-naïve Parkinson’s disease
Seong Ho Jeong, Eun-Chong Lee, Seok Jong Chung, Hye Sun Lee, Jin Ho Jung, Young H. Sohn, Joon-Kyung Seong, Phil Hyu Lee
npj Parkinson's Disease.2022;[Epub] CrossRef - Effectiveness and safety of electroacupuncture in treating Parkinson disease
Wei Xu, Sha OuYang, Zhenhai Chi, ZhiQin Wang, DaoCheng Zhu, RiXin Chen, GenPing Zhong, FengTing Zhang, GuiQin Zhou, SiWei Duan, Lin Jiao
Medicine.2021; 100(10): e25095. CrossRef - Differences in cause and 12-month follow-up outcome of parkinsonian symptoms in depressed older adults treated with antipsychotics: a case series
Anastasios Politis, Nikolaos Kokras, Michael Souvatzoglou, Kostas Siarkos, Panagiotis Toulas, Constantin Potagas, Theodoros Hatzipanagiotou, Georgios Limouris, Panagiotis Alexopoulos
BMC Psychiatry.2021;[Epub] CrossRef - Effectiveness and safety of moxibustion for Parkinson disease
Yonghui Hou, Baile Ning, Yamin Liu, Ying Liu, Wenbin Fu, Zehuai Wen
Medicine.2021; 100(23): e26256. CrossRef - Glucocerebrosidase Mutations and Motor Reserve in Parkinson’s Disease
Seok Jong Chung, Phil Hyu Lee, Young H. Sohn, Yun Joong Kim
Journal of Parkinson's Disease.2021; 11(4): 1715. CrossRef - Analysis of pupillometer results according to disease stage in patients with Parkinson’s disease
Sooyeoun You, Jeong-Ho Hong, Joonsang Yoo
Scientific Reports.2021;[Epub] CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite- Figure
-




- Related articles
-
- Copper Deficiency Myeloneuropathy in a Patient With Wilson’s Disease
- A Survey of Perspectives on Telemedicine for Patients With Parkinson’s Disease
- Apomorphine Monotherapy for Parkinson’s Disease: A Neglected Option?
- Investigation of the Long-Term Effects of Amantadine Use in Parkinson’s Disease
- Potential Link Between Cognition and Motor Reserve in Patients With Parkinson’s Disease
