 E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 15(1); 2022 > Article
-
Letter to the editor
Dancing Feet Dyskinesia in a Patient with GBA-PD -
Diana A. Olszewska1,2,3
 , Allan McCarthy4, Alexandra I. Soto-Beasley2, Ronald L. Walton2, Owen A. Ross2,3,5*, Tim Lynch1,2*
, Allan McCarthy4, Alexandra I. Soto-Beasley2, Ronald L. Walton2, Owen A. Ross2,3,5*, Tim Lynch1,2* -
Journal of Movement Disorders 2022;15(1):83-85.
DOI: https://doi.org/10.14802/jmd.20169
Published online: May 3, 2021
1The Dublin Neurological Institute at the Mater Misericordiae University Hospital, Dublin, Ireland
2Department of Neuroscience, Mayo Clinic, Jacksonville, FL, USA
3School of Medicine and Medical Science, University College Dublin, Dublin, Ireland
4Department of Neurology, The Adelaide and Meath Hospital, Dublin, Ireland
5Department of Clinical Genomics, Mayo Clinic, Jacksonville, FL, USA
- Corresponding author: Diana A. Olszewska, MD, PhD The Dublin Neurological Institute at the Mater Misericordiae University Hospital, 57 Eccles Street, Dublin, Ireland / Tel: +0035318545031 / E-mail: diana.angelika.olszewska@gmail.com
- *These authors contributed equally to this work.
Copyright © 2022 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 4,372 Views
- 154 Download
- 1 Web of Science
- A 70-year-old Irish woman was diagnosed with EOPD at age 49 shortly after developing right-hand bradykinesia and rigidity. There was no family history of note. She commenced levodopa/carbidopa/entacapone 100/25/200 mg four times/day (QID) [levodopa equivalent daily dose (LEDD) 532]. At age 51 years, she developed symmetrical dancing-like lower limb dyskinesia. Dyskinesia was reported to occur within 30 minutes of levodopa administration, last for 30 minutes, and recur 30 minutes before the next dose, suggesting a diphasic nature; therefore, the therapy was changed to levodopa/carbidopa 100/25 mg, and the dose was increased to two tablets three times/day (TID) (LEDD 600), which resulted in a worsening of the dyskinesia. Over the years, different strategies were tried, including the addition of amantadine 100 mg TID without benefit (LEDD 832) (discontinued). At age 67, while on levodopa/carbidopa two tablets TID, she had to sit down during the episodes of dyskinesia, resulting in a tendency to fall off the sofa (Supplementary Video 1 in the online-only Data Supplement). She used a walker, as her balance was severely affected by dyskinesia. A benefit was seen after the individual doses were decreased and the frequency was increased, from 2 tablets TID to 1.5 tablets QID (LEDD 600). At age 68, to improve the continuity of the dopaminergic delivery, a rotigotine patch was tried; however, it was discontinued (nausea, confusion). Dyskinesia, right-foot dystonia, and severe freezing of gait contributed to balance and gait difficulties, resulting in falls/fractures. Subsequently, levodopa/carbidopa was decreased to 100/25 mg 1 tablet QID and 1.5 tablets once/day (LEDD 550 mg), with further improvement (able to use a cane, no falls, did not have to sit down during dyskinesia). She developed hallucinations at age 63 years (transiently resolved with quetiapine 25 mg) and dementia at age 69 years (Montreal Cognitive Assessment test score 14/30). Currently (age 72), she is a nursing-home resident on levodopa/carbidopa 100/25 mg 5 times/day and a slow-release formulation at bedtime (LEDD 575). On 500 mg during the day, the daytime dyskinesia resolved and occurred solely as an end-of-dose evening phenomenon.
- On examination (age 70, LEDD 550 mg, 1-hour post levodopa), there was slight neck rigidity, bradykinesia in both the upper (mild) and lower limbs (slight), severe retropulsion and dystonic posturing of both feet. There was no tremor or upper limb dyskinesia; however, dancing-like lower limb dyskinesia was present bilaterally (Supplementary Video 2 in the online-only Data Supplement). The MDS-Unified Parkinson’s Disease Rating Scale (MDS-UPDRS)-I score was 9, II:31, III:24, and IV:3. The Hoehn and Yahr (H&Y) scale score was 3.
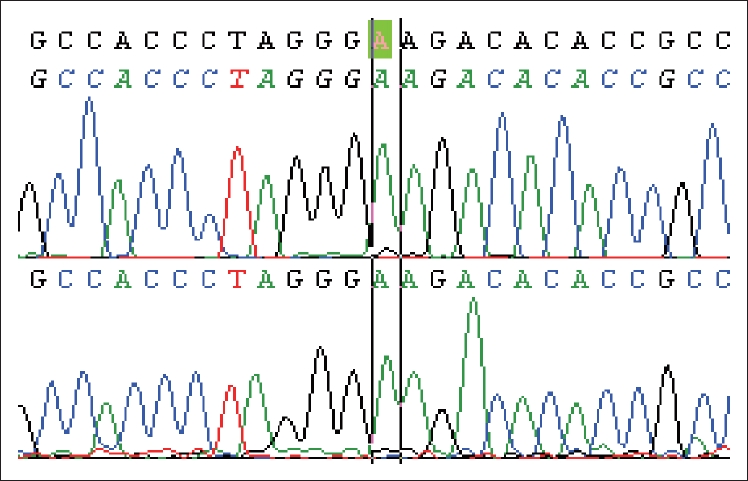
- Genetic testing for parkin, PINK1, and DJ1 variants (full Sanger sequencing, multiplex ligation-dependent probe amplification analysis) was negative. Further testing revealed a missense homozygous GBA exon 8 variant p.Glu365Lys (c.1093 G>A, E326K/E326K) (Figure 1).
CASE
- While parkin gene pathogenic variants are associated with symmetrical dopaminergic neuronal loss (pigmented neuronal loss, bilateral gliosis in the ventrolateral substantia nigra and more symmetric dopaminergic striatal innervation loss with a slower progression rate on dopamine transporter single-photon emission computerized tomography scan) [1,4], the deficit in GBA-PD is more asymmetrical, similar to that seen in IPD/LRRK2-PD [4]. The dancing feet phenomenon in parkin-PD was described in 2012 by Chang et al., who hypothesized that the symmetrical loss of neurons in parkin-PD could be responsible for the symmetrical pattern of lower limb dyskinesia [1]. The presence of this phenomenon in a GBA carrier may indicate a different yet unknown mechanism. Interestingly, E326K/E326K variants do not cause Gaucher’s disease and are rarely reported in a homozygote state [2].
- While lower limb dyskinesia is not specific to any particular type of dyskinesia, it occurs more frequently in diphasic dyskinesia. Diphasic dyskinesia usually appears within 30 minutes of levodopa administration and then subsides and recurs within an hour of the next dose (in keeping with the early description in our patient) [5,6]. Different strategies were attempted to address dyskinesia in our patient, including a levodopa dose increase (reported to improve diphasic dyskinesia), which failed to benefit [5,6]. The initial change from levodopa/carbidopa/COMTI to levodopa/carbidopa alone resulted in a slight improvement. The greatest benefit occurred with a decrease in the individual dose and an increase in the frequency (more in keeping with a peak dose dyskinesia) [5,6]. Based on the patient’s observation (age 70), we concluded that there was peak dose dyskinesia at that point; however, diphasic dyskinesia may be mistaken for peak dose dyskinesia, especially at the lowest dose, when diphasic dyskinesia becomes dominant between levodopa doses [6]. This conundrum could be solved by apomorphine use or deep brain stimulation surgery; however, our patient was not suitable for these treatments at the later stage of her disease (dementia, hallucinations, dopamine agonist intolerance) [6,7].
- With phenotypic overlap, reliable clinical genotype-phenotype correlations in PD are currently not possible. Dyskinesia occurs more frequently in EOPD, regardless of the genotype; however, the combination of an early age-at-onset, excellent response to low levodopa doses and early motor fluctuation development may aid the decision to proceed with genetic testing.
- Our new observation emphasizes that the dancing feet phenomenon early in the course of EOPD may indicate the diagnosis of not only parkin-PD but also GBA-PD when the progression is faster and cognitive symptoms are present. Such patients should be considered for GBA testing, which is not included in the EOPD panel. The inclusion of GBA testing in the EOPD genetic test panel should be considered.
DISCUSSION
Supplementary Video Legends
Video 1.
Video 2.
-
Ethics Statement
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Institutional Research Committee (Mater Misericordiae University Hospital, Dublin, Ireland, ethical approval number 1/378/1300) and with the 1975 Declaration of Helsinki and its later amendments or comparable ethical standards. Informed consent was obtained from the patient included in the study.
-
Conflicts of Interest
The authors have no financial conflicts of interest.
-
Author Contributions
Conceptualization: Diana A. Olszewska, Owen A. Ross, Tim Lynch. Data curation: all authors. Formal analysis: all authors. Investigation: all authors. Methodology: all authors. Project administration: all authors. Resources: all authors. Software: Diana A. Olszewska, Alexandra I. Soto-Beasley, Ronald L. Walton Supervision: Owen A. Ross, Tim Lynch. Validation: Diana A. Olszewska, Alexandra I. Soto-Beasley, Ronald L. Walton. Visualization: Diana A. Olszewska. Writing—original draft: Diana A. Olszewska. Writing—review & editing: all authors.
Notes
- Owen A. Ross is supported by the National Institutes of Health (NIH; R01NS78086; U54 NS100693; U54 NS110435), the US Department of Defense (W81XWH-17-1-0249), the Little Family Foundation, the Mayo Clinic Center for Individualized Medicine, and the Michael J. Fox Foundation.
- Tim Lynch is supported by the Health Research Board and the Michael J. Fox Foundation.
Acknowledgments
- 1. Chang FC, Mehta P, Koentjoro B, Latt M, Blair N, Nicholson G, et al. “Dancing Feet Dyskinesias”: a clue to parkin gene mutations. Mov Disord 2012;27:587–588.ArticlePubMed
- 2. Kasten M, Hartmann C, Hampf J, Schaake S, Westenberger A, Vollstedt EJ, et al. Genotype-phenotype relations for the Parkinson’s disease genes parkin, PINK1, DJ1: MDSGene systematic review. Mov Disord 2018;33:730–741.ArticlePubMed
- 3. Jesús S, Huertas I, Bernal-Bernal I, Bonilla-Toribio M, Cáceres-Redondo MT, Vargas-González L, et al. GBA variants influence motor and nonmotor features of Parkinson’s disease. PLoS One 2016;11:e0167749. ArticlePubMedPMC
- 4. McNeill A, Wu RM, Tzen KY, Aguiar PC, Arbelo JM, Barone P, et al. Dopaminergic neuronal imaging in genetic Parkinson’s disease: insights into pathogenesis. PLoS One 2013;8:e69190.ArticlePubMedPMC
- 5. Vijayakumar D, Jankovic J. Drug-induced dyskinesia, Part 1: treatment of levodopa-induced dyskinesia. Drugs 2016;76:759–777.ArticlePubMed
- 6. Verhagen Metman L, Espay AJ. Teaching video NeuroImages: the underrecognized diphasic dyskinesia of Parkinson disease. Neurology 2017;89:e83–e84.ArticlePubMedPMC
- 7. Kim A, Kim HJ, Kim A, Kim Y, Jang M, Paek SH, et al. Bilateral subthalamic nucleus deep brain stimulation is an effective treatment for diphasic dyskinesia. Eur J Neurol 2021;Jan. 29. [epub]. https://doi.org/10.1111/ene.14756. Article
REFERENCES
Figure & Data
References
Citations
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite
