Dear Editor,
Parkinsonism is the first clinical manifestation in juvenile-onset Huntington’s disease (HD) and in some adult or late-onset HD patients [1]. Even in adult-onset HD, chorea in the early stage of the disease is gradually replaced by akinetic rigid symptoms in the late stage of the disease. Although the degeneration of postsynaptic striatal neurons participating in the direct pathway is considered to be a key mechanism of parkinsonism [2], a contribution of presynaptic dopaminergic dysfunction was suggested by molecular imaging studies targeting nigrostriatal presynaptic terminals in adult-onset HD patients presenting with parkinsonism or even typical chorea [1,3,4]. Here, we report an additional adult-onset HD patient presenting with slowly progressive parkinsonism who exhibited severe striatal atrophy and hypometabolism in contrast to relatively preserved nigrostriatal input.
A 33-year-old male patient first visited our movement disorders clinic for slowness of body movement. He had grown up normally without any physical or intellectual problems until he first felt a very mild postural tremor and unsteady walking in his early twenties. Nevertheless, he was able to complete his mandatory military service without any problems. His symptoms slowly progressed to become noticeable by friends in his mid-20s, and he started to feel clumsiness in his hands, voice tremor, and cognitive decline at age 29. He was born a second child of two half-siblings with a common father. His 71-year-old father and 45-year-old half-sister with a common father did not have neurological illness. His biological mother had been treated for Parkinson’s disease and had showed progressive slowness of movement and gait disturbance since her 30s, and she died at age 45.
On neurological examination, he showed mild masked face and mild voice tremor, but rest tremor and dyskinesia were not visible on limbs. When he stretched his arms out, he showed a fine postural tremor on both hands and a hyperextending dystonic posture on the left fingers. A moderate degree of rigidity was observed on his arms, and his legs were mildly spastic. The amplitude of alternating hand movement and finger tapping movement was moderately reduced with a decrement. Rigidity and bradykinesia were more prominent in his left limbs. Limb ataxia was absent. He walked with a mildly short stride and slightly slow speed, while his arm swing was absent. The deep tendon reflex was increased on the left arm, both knees and ankles, and ankle clonus was observed on both sides. His Movement Disorders Society-sponsored Unified Parkinson’s Disease Rating Scale motor score was 46. Neuropsychological test performance indicated impairment in all tested domains, and the Korean version of the Mini-Mental State Examination score was 24.
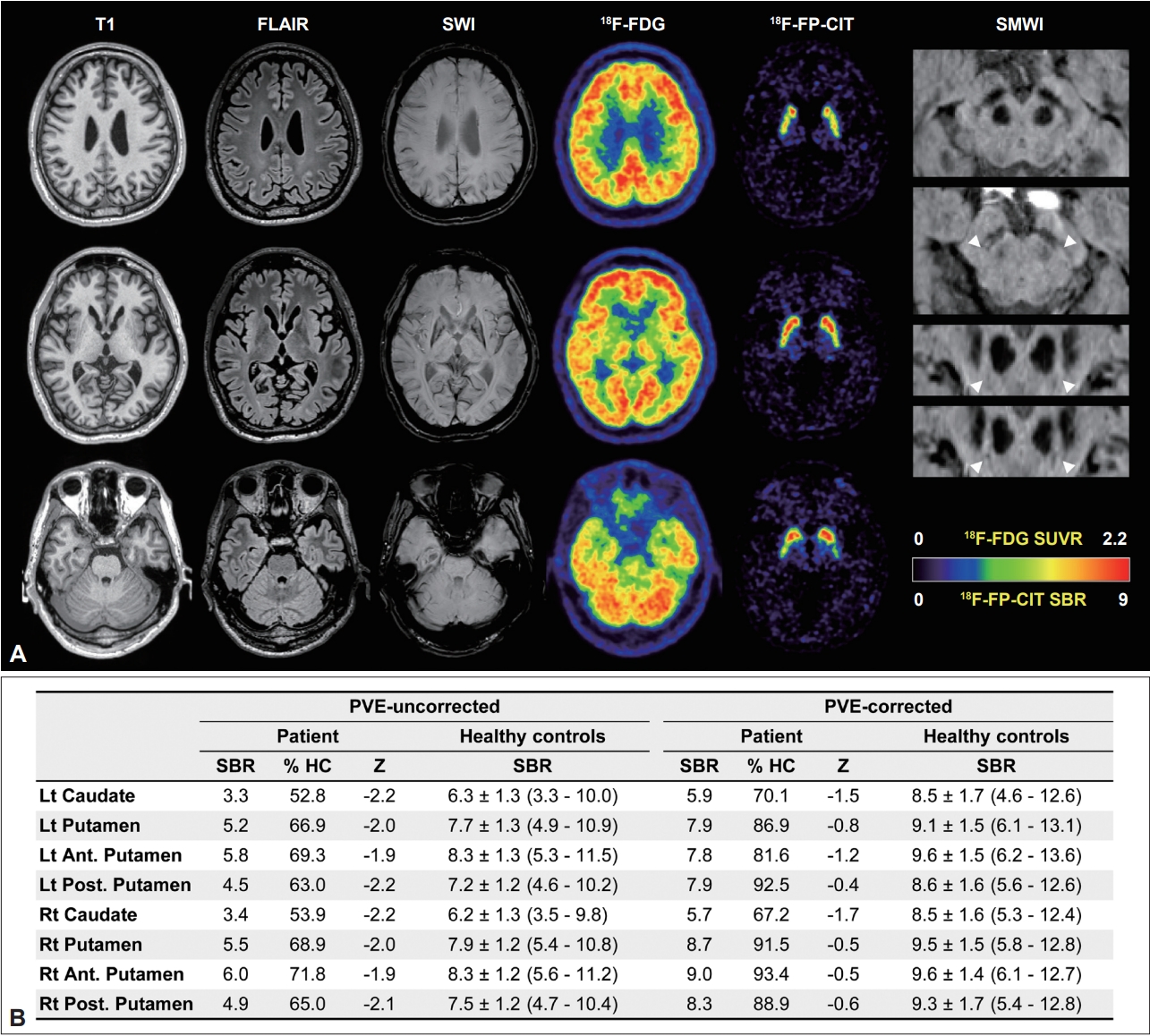
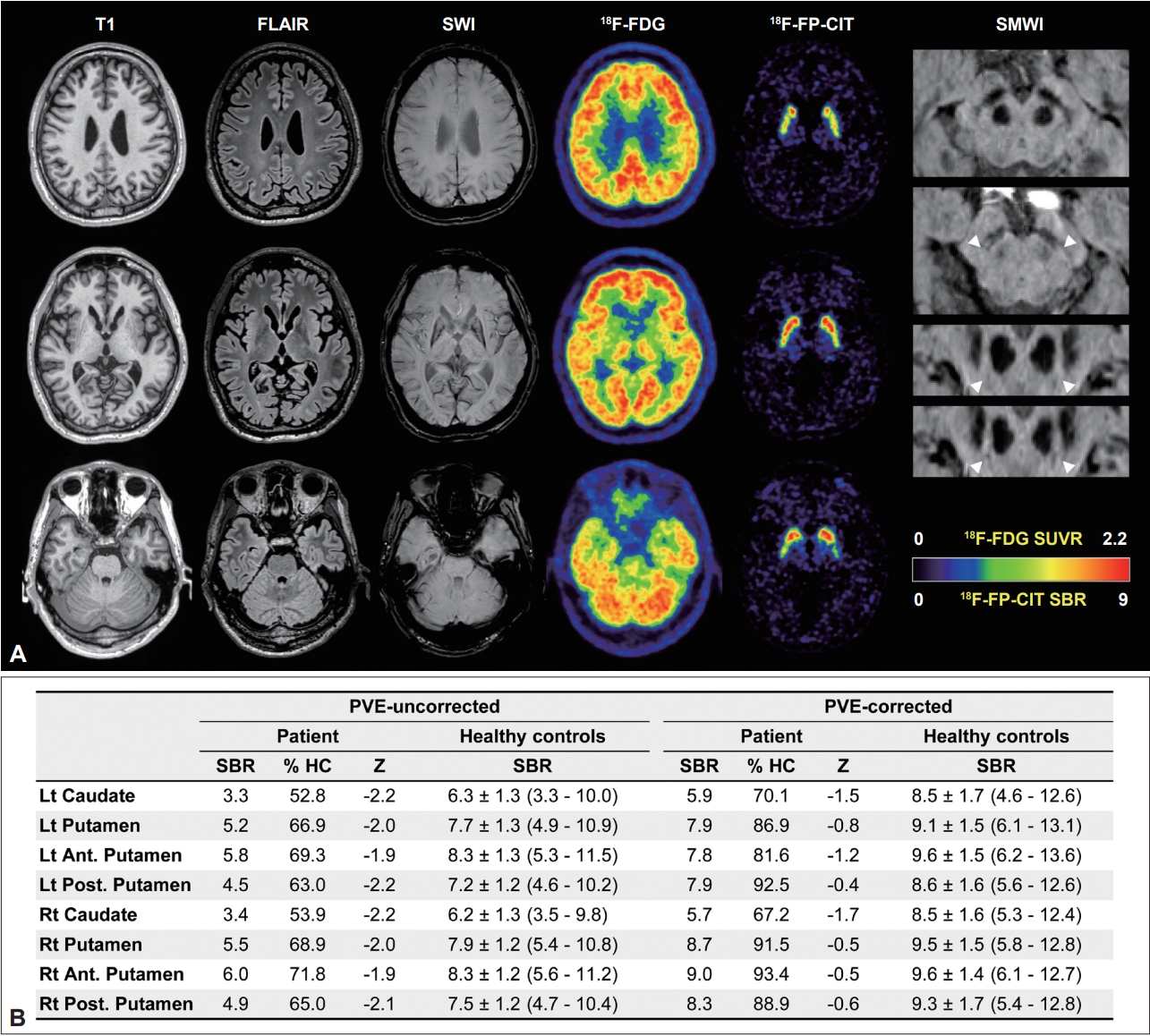
Laboratory tests, including complete blood count, routine chemistry, creatine kinase, and peripheral blood smear, were all normal. A brain magnetic resonance (MR) imaging study exhibited severe atrophy in the entire striatum, in which glucose metabolism was also markedly decreased in the 18F-fluorodeoxyglucose positron emission tomography (PET) study (approximately 50% of the mean standardized uptake value ratio of 71 controls, Z score < -5.0) and in the thalamus. However, cortical metabolism was relatively preserved (Z score > -2.0). Striatal 18F-FP-CIT uptake was mildly decreased in both the putamen (68% of the mean of the controls; Z score, -2.1) and caudate (53% of the mean of the controls; Z score, -2.3). However, when we quantitated striatal uptake after correcting for partial volume effects, the 18F-FPCIT specific binding ratio was almost recovered to the normal range in the putamen (89% of the mean of the controls; Z score, -0.7) and remained at a subnormal level in the caudate (69% of the mean of the controls; Z score, -1.6) (Figure 1 and Supplementary Figure 1 and Supplementary Table 1 in the online-only Data Supplement). Moreover, nigrosome 1 was clearly visible on both sides in the susceptibility map-weighted imaging study (Figure 1). Genetic tests for spinocerebellar ataxia 8 and 17 and dentatorubropallidoluysian atrophy were all normal, but he had a pathologically expanded CAG repeat allele in the Huntington’s disease gene (repeat number 22/52). Although there was only a mild improvement in limb rigidity after treatment with levodopa 300 mg/day, he felt no subjective improvement.
There have been several postmortem reports regarding the involvement of nigral dopaminergic neurons in HD. In contrast, tyrosine hydroxylase-positive nigral neurons were completely unaffected in young-age-onset or juvenile-onset HD patients presenting with predominant parkinsonism [2]. Neuronal loss was observed only in the substantia nigra pars reticulata, in contrast to the normal pigmented neuron number in the shrunken pars compacta in all grades of HD pathology [5]. Moreover, the concentration of striatal vesicular monoamine transporter 2 (VMAT2) measured by autoradiography of postmortem tissue was unchanged or even increased in adult-onset HD patients [6].
Unlike these postmortem studies, imaging studies for striatal dopaminergic input have consistently reported presynaptic dopaminergic deficits. In addition to markedly reduced tracer binding to the striatal D1 and D2 receptors, striatal 11 C-β-CIT uptake was reduced by 50% in adult-onset HD patients [7]. In a 123 I-FP-CIT single photon emission tomography and MR-based volumetry study, half of the HD patients showed reduced putaminal 123 IFP-CIT uptake, and the mean striatal and nigral volumes of the HD patients were smaller than those of the age-matched controls [3]. Likewise, one presynaptic imaging study targeting VMAT2 exhibited a reduction in striatal uptake, particularly in HD patients with predominant parkinsonism [4]. However, tracer binding to striatal VMAT2 was normalized by the partial volume effect [4]. Our patient also exhibited significantly decreased putaminal uptake, only before correcting for the partial volume effect, but almost recovered to the normal range (Z score > -2) after correcting for the partial volume effect. Therefore, the amount of dopaminergic deficit observed in the previous studies was entirely attributable to striatal atrophy or was at least overestimated. Parkinsonian motor dysfunction seems to be much more related to the degeneration of striatal neurons than to nigrostriatal dopaminergic dysfunction.
Nevertheless, there remains an unsolved question regarding levodopa responsiveness in some adult-onset HD patients with predominant parkinsonism and mild loss of nigrostriatal dopaminergic input [1]. Therefore, it seems to be still unclear whether the degeneration of nigral dopaminergic neurons is uniformly observed in HD progression or limited to only some patients.
Supplementary Materials
The online-only Data Supplement is available with this article at https://doi.org/10.14802/jmd.22071.
Supplementary Table 1.
Striatal subregional 18F-FP-CIT uptake values before and after correcting for PVE
jmd-22071-suppl1.pdf
Supplementary Figure 1.
18F-FP-CIT PET images before and after correcting for PVE. Compared to the mean 18F-FP-CIT PET image of the controls, striatal uptake is mildly reduced in the PVE-uncorrected PET image of the HD patient. After correcting for PVE, the 18F-FP-CIT SBR values approach to normal level in the most of the voxels in the atrophic striatum. PVE, partial volume effect; HC, healthy controls; SBR, specific binding ratio; PET, positron emission tomography; HD, Huntington’s disease.
jmd-22071-suppl2.pdf
Notes
-
Ethics Statement
This study was approved by the Institutional Review Board of Gangnam Severance Hospital (IRB number: 3-2022-0071). The requirement of informed consent was waived by the Institutional Review Board.
-
Conflicts of Interest
The authors have no financial conflicts of interest.
-
Funding Statement
This research was supported by a faculty research grant from Yonsei University College of Medicine (6-2021-0094).
-
Author Contributions
Conceptualization: Chul Hyoung Lyoo. Data curation: Taewon Kim, Byeong Joo Choi. Formal analysis: Taewon Kim, Chul Hyoung Lyoo. Funding acquisition: Chul Hyoung Lyoo. Investigation: Taewon Kim. Methodology: Chul Hyoung Lyoo. Project administration: Chul Hyoung Lyoo. Resources: Chul Hyoung Lyoo. Software: Chul Hyoung Lyoo. Supervision: Chul Hyoung Lyoo. Validation: Chul Hyoung Lyoo. Visualization: Chul Hyoung Lyoo. Writing—original draft: Taewon Kim. Writing—review & editing: Chul Hyoung Lyoo.
Figure 1.Imaging studies of patients with Huntington’s disease. In the upper Panel (A), T1-weighted, FLAIR, and SWI MR images show severe atrophy and increased iron content in both the caudate and putamen. Unlike the cerebral cortex showing normal glucose metabolism in the 18F-FDG PET study, markedly reduced glucose metabolism was observed in the entire striatum. Partial volume effect-uncorrected 18F-FP-CIT PET images exhibit mild diffuse reduction of striatal uptake. In contrast, SMWI clearly showed nigrosome 1 (arrowheads) in both substantia nigra. The lower Panel (B) shows the striatal regional 18F-FP-CIT SBR values before and after correcting for the PVE. FLAIR, fluid-attenuated inversion recovery; SWI, susceptibility-weighted; FDG, fluorodeoxyglucose; PET, positron emission tomography; SMWI, susceptibility map-weighted imaging; SUVR, Standardized Uptake Value Ratio; SBR, specific binding ratio; HC, healthy controls; PVE, partial volume effect.
REFERENCES
- 1. Kwak IH, Kim NH, Ma HI, Kim YE. Huntington’s disease presenting as adult-onset parkinsonism. J Clin Neurol 2022;18:87–89.ArticlePubMedPMCPDF
- 2. Albin RL, Reiner A, Anderson KD, Penney JB, Young AB. Striatal and nigral neuron subpopulations in rigid Huntington’s disease: implications for the functional anatomy of chorea and rigidity-akinesia. Ann Neurol 1990;27:357–365.ArticlePubMed
- 3. Kiferle L, Mazzucchi S, Unti E, Pesaresi I, Fabbri S, Nicoletti V, et al. Nigral involvement and nigrostriatal dysfunction in Huntington’s disease: evidences from an MRI and SPECT study. Parkinsonism Relat Disord 2013;19:800–805.ArticlePubMed
- 4. Bohnen NI, Koeppe RA, Meyer P, Ficaro E, Wernette K, Kilbourn MR, et al. Decreased striatal monoaminergic terminals in Huntington disease. Neurology 2000;54:1753–1759.ArticlePubMed
- 5. Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol 1998;57:369–384.ArticlePubMed
- 6. Suzuki M, Desmond TJ, Albin RL, Frey KA. Vesicular neurotransmitter transporters in Huntington’s disease: initial observations and comparison with traditional synaptic markers. Synapse 2001;41:329–336.ArticlePubMed
- 7. Ginovart N, Lundin A, Farde L, Halldin C, Bäckman L, Swahn CG, et al. PET study of the pre- and post-synaptic dopaminergic markers for the neurodegenerative process in Huntington’s disease. Brain 1997;120(Pt 3):503–514.ArticlePubMed
Citations
Citations to this article as recorded by

 E-submission
E-submission
 , Byeong Joo Choi
, Byeong Joo Choi
 PubReader
PubReader ePub Link
ePub Link Cite
Cite