Department of Neurology, Post Graduate Institute of Medical Education and Research, Chandigarh, India
Corresponding author: Sahil Mehta, MD, DM Department of Neurology, Post Graduate Institute of Medical Education and Research, Madhya Marg, Sector 12, Chandigarh 160012, India / Tel: +91-9815543539 / E-mail: mehtasahilpgi@gmail.com
• Received: September 10, 2022 • Revised: October 16, 2022 • Accepted: November 9, 2022
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Developmental and epileptic encephalopathies are rare neurodevelopmental disorders characterized by early onset intractable seizures and frequent epileptiform activity on electroencephalogram associated with developmental delay or regression. The advent of next-generation sequencing has paved the way for discovering new genes responsible for these catastrophic epilepsy syndromes. Hyperkinetic movements have been described in some of these epileptic encephalopathies, such as mutations in ARX, GNAO1, CDKL5 and FOXG1 genes. We report a young female who presented with refractory epilepsy and hyperkinetic movement disorder comprising myoclonus, chorea and ataxia and was found to have a mutation in the DHDDS gene.
A 25-year-old Sikh female born out of nonconsanguineous marriage presented with a history of generalized tonic–clonic convulsions since six months of age. The frequency of these seizures was approximately 3–4 per month. At approximately the same time, the parents also noticed abnormal involuntary body movements. The movements were rapid and jerky, predominantly involving the upper part of the body and face and occasionally leading to falls. Her parents could not describe the exact onset or progression of these movements. They were not associated with any behavioral arrest or loss of consciousness. The seizures were refractory despite multiple antiepileptic drugs (levetiracetam 1.5 g/day, valproate 1 g/day, clobazam 10 mg/day, phenytoin 300 mg/day in various combinations). She was a product of full-term normal vaginal delivery at home with a history of delayed crying at birth but no prenatal complications. Her motor and language milestones were delayed (sitting: 2 years; walking: 6 years; speech [monosyllables]: 2 years of age), and her intelligence quotient was 55, suggestive of mild intellectual disability. Her family history was negative for epilepsy or movement disorder. The parents reported an almost nonprogressive course of her illness.
General physical and systemic examination was noncontributory. Her visual acuity was 6/6 with a normal fundoscopic examination. Extraocular movements revealed impaired upward saccades and deficits in gaze holding. The rest of the cranial nerves were normal. Examination of the motor system was normal except for the presence of hypotonia in the lower limbs. The deep tendon reflexes were 2+. Examination of the hyperkinetic movement disorder revealed generalized action myoclonus, predominantly affecting the upper limbs and face. There was no stimulus sensitivity. There were superimposed choreiform movements, predominantly affecting the distal upper extremities, oro-lingual region and lower limbs. The speech was interrupted due to superimposed myoclonic jerks. She was clumsy on rapid alternating hand movements with past pointing on the left side. Her gait was broad-based and ataxic (Supplementary Video 1 in the online-only Data Supplement).
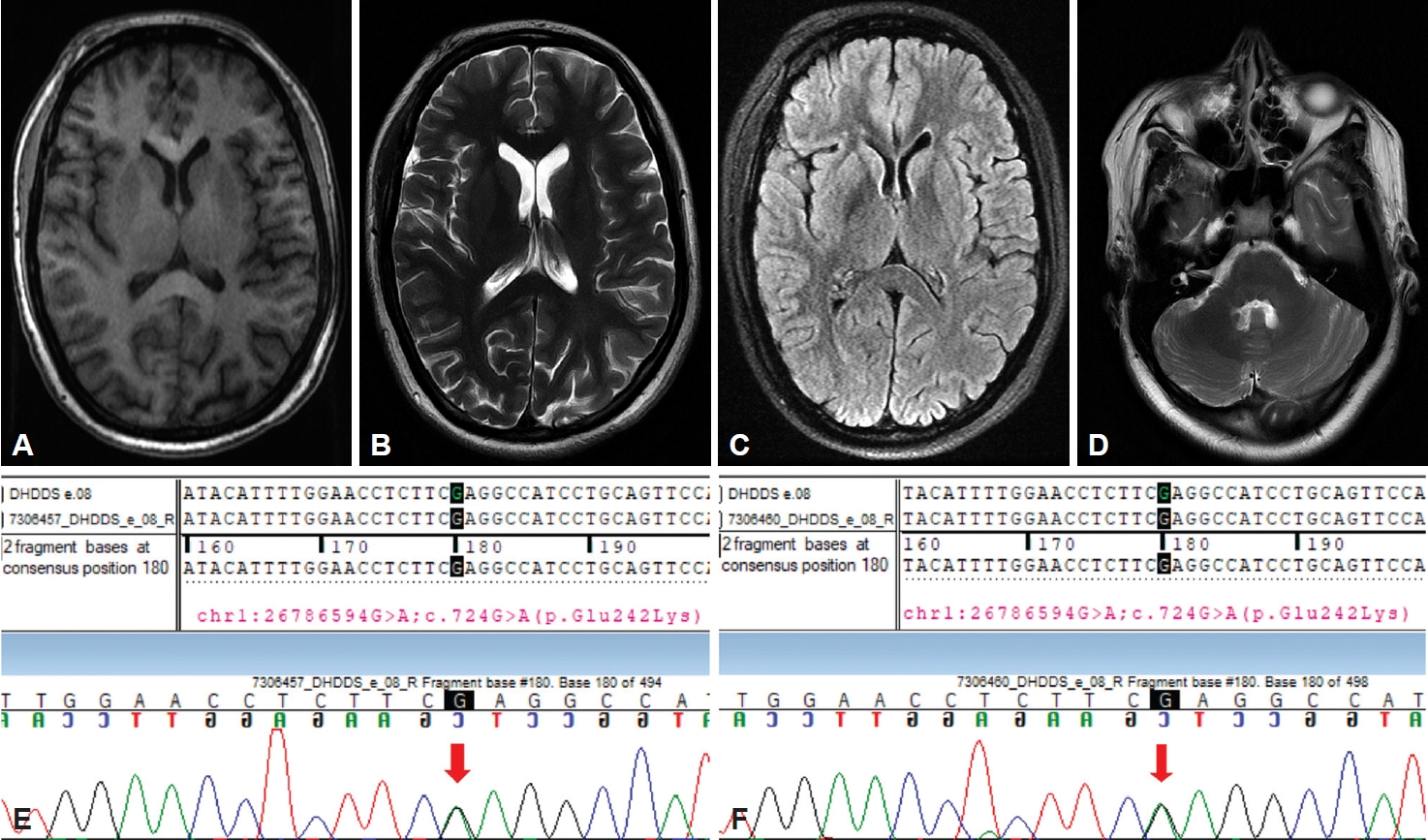
Magnetic resonance imaging brain was noncontributory (Figure 1A–D). Electroencephalogram (EEG) revealed diffuse slowing without any spike-wave discharges. Her routine blood investigations, ceruloplasmin, thyroid profile, antinuclear antibodies, ultrasound whole abdomen and nerve conduction studies were all within normal limits. The possibility of drug-induced movement disorders seemed less likely due to the lack of a significant temporal relationship. Clinical exome sequencing revealed a heterozygous missense variant c.724 G>A (p. Glu242Lys) in exon 8 of the DHDDS gene, confirming the diagnosis of developmental delay and seizures with or without movement abnormalities (DEDSM). The variant was confirmed by Sanger sequencing (Figure 1E) and was classified as likely pathogenic according to American College of Medical Genetics and Genomics guidelines. In silico prediction tools (Polyphen-2, SIFT, mutation taster 2) found this variant to be damaging. Minor allele frequency was found to be low in the general population (0.000% gnomAD south Asian frequency, 0.12% ECGIdb). The exact pathogenic variation was detected in the heterozygous condition in the patient’s father who only had stuttering speech since childhood (Figure 1F). Her father did not suffer from seizures, ataxia or any other movement disorder apart from stammering.
DEDSM is an autosomal dominant neurodevelopmental disorder characterized by global developmental delay, variable intellectual disability, and delayed motor development with early onset seizures [1]. The most characteristic semiology described is generalized epilepsy with myoclonic seizures, which was present in our patient. Myoclonic seizures can be in the form of myoclonic absences or isolated cortical myoclonus and can be precipitated by light or fever. Atonic seizures have also been described. EEG may show generalized spike-wave discharges. A variety of movement disorders have been described in this condition, namely, ataxia, dystonia and tremor. A notable feature is the presence of hypotonia.
DHDDS encodes dehydrodolichyl diphosphate synthase, which is essential for dolichol monophosphate synthesis and global N-linked glycosylation. Homozygous mutations in the DHDDS gene (c.124 A>G) have been associated with nonsyndromic retinitis pigmentosa in consanguineous families.
DHDDS mutations causing a variety of movement disorders have been reported mainly as case series or individual case reports [2-5]. Recently, Galosi et al. [6] and Jiao et al. [7] described 25 and 10 patients, respectively, with this mutation. Myoclonus, either epileptic or nonepileptic, was an important feature in all patients. Tremor and ataxia were present in the majority of the patients, while few had a cortical myoclonic tremor. Parkinsonism, chorea, dystonia and myokymia have also been described. Kim et al. [2] reported a novel variant in the DHDDS gene (c.109C>T, [p. Arg37Cys]) in a patient with adult-onset progressive myoclonus ataxia (Supplementary Material in the online-only Data Supplement). We have summarized all the reported cases in Supplementary Table 1 (in the online-only Data Supplement). Of note, our variant was different from those reported in the literature.
To conclude, mutations in the DHDDS gene should be considered in the differential diagnosis of patients presenting with early onset hyperkinetic movement disorder, especially in the context of developmental delay and epilepsy. The role of next-generation sequencing in identifying rare genetic causes of epileptic encephalopathies cannot be overemphasized.
The video depicts myoclonus predominantly affecting the upper limbs and face with superimposed choreiform movements affecting the distal upper extremities, orolingual region and lower limbs. Speech is interrupted due to superimposed myoclonic jerks. Examination of the oculomotor system shows impaired upward saccades with deficits in gaze holding. Clumsiness is evident on dysdiadochokinesia with past pointing on the left side. Gait is broad-based and ataxic.
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this work is consistent with those guidelines. We also confirm that the patient has given written informed consent for the publication of her video.
Conflicts of Interest
The authors have no financial conflicts of interest.
Magnetic resonance imaging and chromatogram of the patient. (A-D) Depicts normal brain magnetic resonance imaging including basal ganglia and cerebellum. Sequence chromatogram showing the variation in exon 8 of the DHDDS gene in the index patient (E) and her father (F). Arrows denote missense variation in the patient (E) and her father (F).
REFERENCES
1. Hamdan FF, Myers CT, Cossette P, Lemay P, Spiegelman D, Laporte AD, et al. High rate of recurrent de novo mutations in developmental and epileptic encephalopathies. Am J Hum Genet 2017;101:664–685.PubMedPMC
2. Kim J, Kim I, Koh SB. A novel variant of dehydrodolichol diphosphate synthase (DHDDS) mutation with adult-onset progressive myoclonus ataxia. Parkinsonism Relat Disord 2021;87:135–136.ArticlePubMed
3. Wood K, Montgomery T, Devlin AM. DHDDS related epilepsy--report of familial cases and review of the literature. Seizure 2021;91:189–191.ArticlePubMed
4. Kim S, Kim MJ, Son H, Hwang S, Kang MK, Chu K, et al. Adult-onset rapidly worsening progressive myoclonic epilepsy caused by a novel variant in DHDDS. Ann Clin Transl Neurol 2021;8:2319–2326.ArticlePubMedPMCPDF
5. Courage C, Oliver KL, Park EJ, Cameron JM, Grabińska KA, Muona M, et al. Progressive myoclonus epilepsies-residual unsolved cases have marked genetic heterogeneity including dolichol-dependent protein glycosylation pathway genes. Am J Hum Genet 2021;108:722–738.ArticlePubMedPMC
6. Galosi S, Edani BH, Martinelli S, Hansikova H, Eklund EA, Caputi C, et al. De novo DHDDS variants cause a neurodevelopmental and neurodegenerative disorder with myoclonus. Brain 2022;145:208–223.ArticlePubMedPMCPDF
7. Jiao X, Xue Y, Yang S, Gong P, Niu Y, Wang Q, et al. Phenotype of heterozygous variants of dehydrodolichol diphosphate synthase. Dev Med Child Neurol 2022;64:125–134.ArticlePubMedPDF
Figure & Data
References
Citations
Citations to this article as recorded by
DHDDS and NUS1: A Converging Pathway and Common Phenotype Laura J. Williams, Sophie Waller, Jessica Qiu, Emily Innes, Noha Elserafy, Peter Procopis, Hugo Sampaio, Neil Mahant, Michel C. Tchan, Shekeeb S. Mohammad, Hugo Morales‐Briceño, Victor S.C. Fung Movement Disorders Clinical Practice.2024; 11(1): 76. CrossRef
 E-submission
E-submission

 , Vivek Lal
, Vivek Lal
 PubReader
PubReader ePub Link
ePub Link Cite
Cite
