E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 16(2); 2023 > Article
-
Case Report
Novel Compound Heterozygous Mutations in the SYNE1 Gene in a Taiwanese Family: A Case Report and Literature Review -
Chia-Yan Kuo1
 , Pei Shan Yu2, Chih-Ying Chao2, Chun-Chieh Wang2, Wen-Lang Fan3,4, Yih-Ru Wu1,2
, Pei Shan Yu2, Chih-Ying Chao2, Chun-Chieh Wang2, Wen-Lang Fan3,4, Yih-Ru Wu1,2
-
Journal of Movement Disorders 2023;16(2):202-206.
DOI: https://doi.org/10.14802/jmd.22105
Published online: April 26, 2023
1Chang Gung University, College of Medicine, Tauyuan, Taiwan
2Department of Neurology, Chang Gung Memorial Hospital, Linkou Medical Center, Tauyuan, Taiwan
3Department of Medical Research, Kaohsiung Chang Gung Memorial Hospital, Kaohsiung, Taiwan
4Genomic Medicine Core Laboratory, Chang Gung Memorial Hospital, Linkou Medical Center, Taoyuan, Taiwan
- Corresponding author: Yih-Ru Wu, MD Department of Neurology, Chang Gung Memorial Hospital, Linkou Medical Center, No 5, Fuxing St. Guishan Dist., Tauyuan 333, Taiwan / Tel: +886-3-3281200-8349 / Fax: +886-3-3287226 / E-mail: yihruwu@cgmh.org.tw
Copyright © 2023 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 1,182 Views
- 86 Download
ABSTRACT
- Mutations in the synaptic nuclear envelope protein 1 (SYNE1) gene are associated with substantial clinical heterogeneity. Here, we report the first case of SYNE1 ataxia in Taiwan due to two novel truncating mutations. Our patient, a 53-year-old female, exhibited pure cerebellar ataxia with c.1922del in exon 18 and c. C3883T mutations in exon 31. Previous studies have indicated that the prevalence of SYNE1 ataxia among East Asian populations is low. In this study, we identified 27 cases of SYNE1 ataxia from 22 families in East Asia. Of the 28 patients recruited in this study (including our patient), 10 exhibited pure cerebellar ataxia, and 18 exhibited ataxia plus syndromes. We could not find an exact correlation between genotypes and phenotypes. Additionally, we established a precise molecular diagnosis in our patient’s family and extended the findings on the ethnic, phenotypic, and genotypic diversity of the SYNE1 mutational spectrum.
- Patients and their clinical characteristics
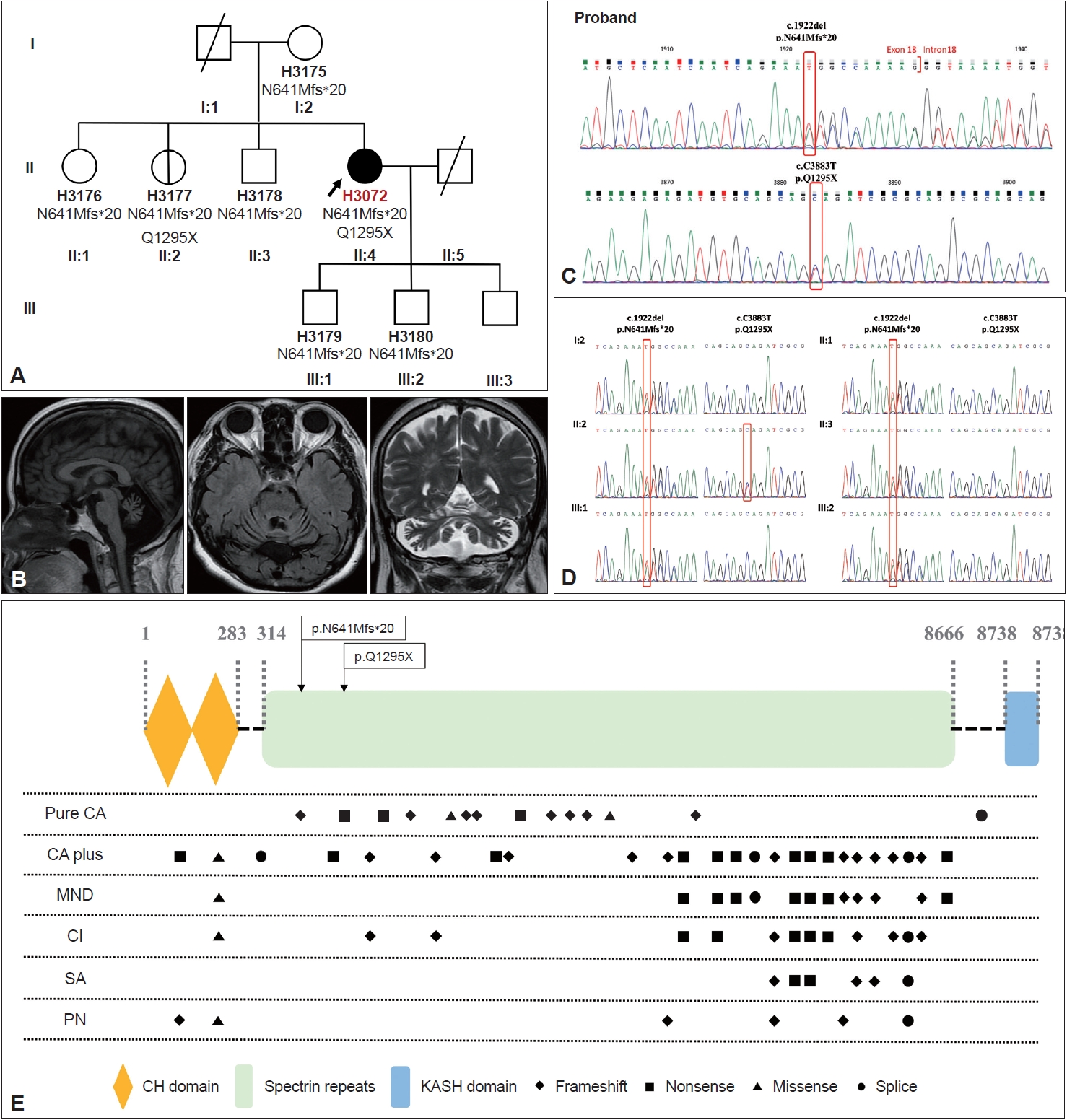
- Our patient was a 53-year-old female from a nonconsanguineous family with healthy parents and older siblings (Figure 1A). She sought care at our hospital because of progressive unsteady gait, slurred speech, and uncoordinated movements of greater than 20 years duration.
- Neurological examination revealed pure cerebellar signs, including gaze-evoked nystagmus, saccadic pursuit with hypometric saccades, scanning speech, dysmetria, dysdiadochokinesia, and wide-based gait. Cranial nerve examination and mini-mental state examination were both normal. Nerve conduction velocity, electromyography, and electroencephalography examinations were normal. Brain magnetic resonance imaging revealed diffuse cerebellar atrophy (Figure 1B).
- Novel heterozygous mutations identified by genomic analysis
- We first extracted genomic DNA from our index patient and then obtained additional samples from unaffected families for segregation analysis. Whole exome sequencing (WES) and parallel sequencing were performed, followed by alignment and mapping to the reference genome. We filtered the mutations from the variations in dbSNP150, HapMap, the 1000 Genomes Project, and the Genome Aggregation Database. Algorithm with ANNOVAR and the VarSome database (https://varsome. com/, accessed on February 25, 2022) were used to verify the pathogenicity of the mutations. Finally, mutations were confirmed using Sanger sequencing. The steps, including the practical tools and databases, are detailed in the Supplementary Materials 1–3 in the online-only Data Supplement.
- Novel heterozygous mutations were identified in exon 18 (NM_182961, c.1922del, and p. N641Mfs) and exon 31 (NM_182961, c. C3883T, and p. Q1295X), which caused frameshift and nonsense mutations (stop-gain) (Figure 1C). These mutations were confirmed using Sanger sequencing. According to the Sanger sequencing results, segregation of the novel SYNE1 variant (c.1922del, p. N641Mfs) was present in all of the patient’s family members and that of the other novel truncating variants (c. C3883T, p. Q1295X) was present in one of her older sisters (II:2) (Figure 1A and D). Only very mild wide-based gait was detected in the index patient. Because of personal issues, she declined to undergo further examinations. These variants have not been previously reported in either the literature or the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php, accessed on May 14, 2022).
- Pathogenicity assessment of SYNE1 variants
- According to the guidelines of the American College of Medical Genetics and Genomics (ACMG) [4], pathogenicity is categorized as follows: very strong evidence of pathogenicity (PVS), strong evidence of pathogenicity (PS), moderate evidence of pathogenicity (PM), and supporting evidence of pathogenicity (PP). Thus, the nonsense mutation c. C3883T is categorized as pathogenic because it satisfies the following criteria: 1) PVS1, 2) PM2, 3) PM3, and 4) PP3. Similarly, the other frameshift mutation, c.1922del, is also categorized as pathogenic because it satisfies the same criteria.
CASE REPORT
- Differential diagnosis
- Our patient exhibited pure cerebellar disorders with no clear noncerebellar signs. Therefore, to establish a diagnosis, we first assessed her history in detail and performed some laboratory tests. Her medical history eliminated the possibility of trauma, infection, toxicity, primary or metastatic tumor, excess alcohol consumption, vitamin deficiency, and immune-mediated disorders that may cause secondary ataxia. The laboratory results, including kidney and liver function tests, revealed normal laboratory findings. Based on the patient’s clinical features (late onset at nearly 30 years of age with a slowly progressing pattern of nearly 20 years duration), we first considered a diagnosis of sporadic spinocerebellar ataxia (SCA). We used a gene panel to screen for CAG-repeat expansion SCA, but the results were negative. We did not test for Friedreich’s ataxia because this disease is extremely rare in the East Asian population. Therefore, we used WES and identified the SYNE1 mutations associated with ARCA. Given the patient’s clinical manifestations, a diagnosis of ARCA1 was finally established.
- Literature review
- ARCA1 is a slowly progressive and relatively pure form of cerebellar ataxia [1]. However, the phenotypic spectrum of this neurodegenerative disorder involves not only cerebellar ataxia but also ataxia plus syndromes with high heterogeneity [3,5]. In a 2016 cohort screening of 434 non-Canadian index patients with autosomal recessive cerebellar ataxia collected from seven centers across Europe, Synofzik et al. [3] reported on the prevalence of cerebellar ataxia plus syndromes (80.8%) with additional system dysfunction and diverse clinical manifestations. Non-Asian populations (Supplementary Tables 2 and 3 in the online-only Data Supplement) have a reportedly high ratio of ataxia plus syndromes in patients with ARCA1. The most common extracerebellar signs of the 26 patients with ARCA1 included in their study were motor neuron dysfunction (57.7%) and skeletal abnormalities (34.6%). Other signs, such as dystonia, reduced vibratory sensation, intellectual disability, dysphagia, respiratory dysfunction, peripheral neuropathy, and Chilaiditi syndromes, have also been reported [3].
- In East Asia, the prevalence of ARCA1 is approximately 1.6%–3.8% in suspected cases of ARCA [6]. In addition, the prevalence of cerebellar ataxia plus syndromes is high, accounting for 18 out of 28 patients (64.3%) from 23 unrelated families. Data regarding the mutations and associated phenotypes of all these East Asian cases, including our patient, are listed in Supplementary Table 1 in the online-only Data Supplement. Among East Asian populations, the median age at onset is 20 years. In our study, the age of onset was earlier in patients with ataxia plus syndromes than in patients with pure ataxia (Supplementary Table 4 in the online-only Data Supplement). Nevertheless, further evidence is needed to confirm a significant difference in clinical manifestations between the two groups due to the small number of currently recognized East Asian cases. In a certain subgroup (21.4%) of Asian patients, SYNE1 disease starts with noncerebellar features [6,7], which have a reportedly incidence of approximately 50% among Caucasian populations [5]. Among Asian patients with ataxia plus syndromes, the main noncerebellar signs include motor neuron disease, cognitive impairment, skeletal and soft-tissue abnormalities, peripheral neuropathy, and intellectual disability (Supplementary Fig. 1 in the online-only Data Supplement).
- Relationship between mutation and phenotype
- The SYNE1 gene, located on chromosome 6, comprises 147 exons, encodes 27,652-kb mRNA and expresses Nesprin-1, a huge protein consisting of 8,797 amino acids. In a previous study, Gros-Louis et al. [1] detected the greatest expression of SYNE1 in the cell bodies of Purkinje cells in the cerebellar cortex and in neurons from the olivary region of the brain stem by using immunohistochemistry staining of normal mouse brain crosssections. Nesprins, a family of intracellular scaffold proteins, serve as anchors among the nuclear envelope, other subcellular compartments, and cytoskeleton [2]. Numerous Nesprin-1 isoforms, generated by alternative initiation of transcription and mRNA splicing, have been identified thus far. Among these isoforms, Nesprin-1-giant is specifically expressed in the central nervous system, with the highest expression level in the cerebellum [8]. In the absence of a KASH domain, Nesprin-1-giant is called KASH-less nesprin-1 giant (KLNes1g), which is colocalized with mossy fiber glomeruli [8]. Loss-of-function mutations in KLNes1g cannot be reverted by Nesprin-2 because of their different biological and cellular functions [8]. As long as ARCA1 mutations, including p. N641Mfs*20 and p. Q1295X, cause KLNes1g truncation, they are usually predicted to underlie the cerebellar phenotype [8].
- To explore the relationship between genotypes and phenotypes, we summarized all cases with a SYNE1 variant-related phenotype in East Asia (Supplementary Table 1 in the onlineonly Data Supplement). According to Indelicato et al. [9], mutations associated with a complicated phenotype are located near the C-terminus, whereas mutations associated with pure cerebellar ataxia are located near the N-terminus. In our study, a similar association was also observed among all 37 ARCA1-related mutations identified in the East Asian population, except for the KASH domain (Figure 1E). Moreover, Baumann et al. [10] reported that ARCA1-associated truncating variants affect the giant and large KLNes1g but leave nesprin-1 alpha 2 intact, while loss of the KASH domain is sufficient to cause SYNE1-associated arthrogryposis multiplex congenita. These genotype–phenotype correlation can be partly explained by the fact that nesprin-1 alpha 2 (120 kDa) represents a C-terminal, predominantly muscle-specific isoform, whereas KLNes1g represents a large KASH-less variant specifically expressed in the CNS and most abundant in the cerebellum [10].
- Based on the idea that Nesprin-1 isoforms and tissue-specific scaffolds might affect clinical manifestations, we performed Basic Local Alignment Search Tool (BLAST) on The Universal Protein Resource (UniProt, https://www.uniprot.org/, accessed on September 17, 2022) and analyzed all the available 33 protein sequences of SYNE1 variants in East Asia except the splice site mutations (the detailed steps are recorded in Supplementary Materials 1–3 in the online-only Data Supplement) to understand genotype and phenotype correlations. The results showed that the SYNE1 variants that were identified in patients with pure cerebellar ataxia predominantly affect N-terminal isoforms. In contrast, the variants that were identified in patients with ataxia predominantly involve C-terminus isoforms (Supplementary Tables 5 and 6, and Supplementary Figure 2 in the online-only Data Supplement). However, with limited information about the structure and function of the Nesprin-1 isoforms, our study cannot explain the diverse spectrum of the disease. Overall, much about the relationship between mutation and phenotype remains to be explained.
- In conclusion, in this study, we identified the first known case of SNYE1-related ataxia in the Taiwanese population due to novel mutations. We also expanded on existing findings regarding the ethnic and genetic diversity of SNYE1-related ataxia. However, the pathological mechanism of SNYE1 mutations remains unknown.
DISCUSSION
Supplementary Material
Supplementary Material 1.
Supplementary Material 2.
Supplementary Material 3.
Supplementary Table 1.
Supplementary Table 2.
Supplementary Table 3.
Supplementary Table 4.
Supplementary Table 5.
Supplementary Table 6.
Supplementary Figure 1.
Supplementary Figure 2.
-
Ethics Statement
We obtained informed consent for genetic testing and case reports from the patient, and we performed a genetic analysis to test for hereditary ataxia (IRB No 202200285B0A3 at Chang Gung Memorial Hospital, Linkou Medical Center). The authors certify that this article complies with the Principles of Ethical Publishing of Journal of Movement Disorders and declare that they acted in accordance with ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
-
Conflicts of Interest
The authors have no financial conflicts of interest.
-
Funding Statement
This work was supported by Minister of Science and Technology, Taiwan (MOST 109-2314-B-182A-077-MY3) and Chang Gung Memorial Hospital, Taipei, Taiwan (CMRPG3K1481).
-
Author contributions
Conceptualization: Yih-Ru Wu. Data curation: Pei Shan Yu. Formal analysis: Chih-Ying Chao, Chun-Chieh Wang, Wen-Lang Fan. Funding acquisition: Yih-Ru Wu. Investigation: Yih-Ru Wu, Pei Shan Yu. Methodology: Chih- Ying Chao, Chun-Chieh Wang, Wen-Lang Fan, Yih-Ru Wu. Project administration: Yih-Ru Wu. Resources: Yih-Ru Wu. Software: Pei Shan Yu, Wen-Lang Fan. Supervision: Yih-Ru Wu. Validation: Chih-Ying Chao, Chun- Chieh Wang, Wen-Lang Fan. Visualization: Yih-Ru Wu. Writing—original draft: Chia-Yan Kuo, Pei Shan Yu. Writing—review & editing: Yih-Ru Wu.
Notes
- 1. Gros-Louis F, Dupré N, Dion P, Fox MA, Laurent S, Verreault S, et al. Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat Genet 2007;39:80–85.ArticlePubMedPDF
- 2. Rajgor D, Shanahan CM. Nesprins: from the nuclear envelope and beyond. Expert Rev Mol Med 2013;15:e5.ArticlePubMedPMC
- 3. Synofzik M, Smets K, Mallaret M, Di Bella D, Gallenmüller C, Baets J, et al. SYNE1 ataxia is a common recessive ataxia with major non-cerebellar features: a large multi-centre study. Brain 2016;139(Pt 5):1378–1393.ArticlePubMedPMC
- 4. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424.ArticlePubMedPMCPDF
- 5. Mademan I, Harmuth F, Giordano I, Timmann D, Magri S, Deconinck T, et al. Multisystemic SYNE1 ataxia: confirming the high frequency and extending the mutational and phenotypic spectrum. Brain 2016;139(Pt 8):e46.ArticlePubMedPMC
- 6. Duan X, Hao Y, Cao Z, Zhou C, Zhang J, Wang R, et al. Autosomal recessive cerebellar ataxia type 1: phenotypic and genetic correlation in a cohort of Chinese patients with SYNE1 variants. Cerebellum 2021;20:74–82.ArticlePubMedPDF
- 7. Peng Y, Ye W, Chen Z, Peng H, Wang P, Hou X, et al. Identifying SYNE1 ataxia with novel mutations in a Chinese population. Front Neurol 2018;9:1111.ArticlePubMedPMC
- 8. Razafsky D, Hodzic D. A variant of Nesprin1 giant devoid of KASH domain underlies the molecular etiology of autosomal recessive cerebellar ataxia type I. Neurobiol Dis 2015;78:57–67.ArticlePubMedPMC
- 9. Indelicato E, Nachbauer W, Fauth C, Krabichler B, Schossig A, Eigentler A, et al. SYNE1-ataxia: novel genotypic and phenotypic findings. Parkinsonism Relat Disord 2019;62:210–214.ArticlePubMed
- 10. Baumann M, Steichen-Gersdorf E, Krabichler B, Petersen BS, Weber U, Schmidt WM, et al. Homozygous SYNE1 mutation causes congenital onset of muscular weakness with distal arthrogryposis: a genotype-phenotype correlation. Eur J Hum Genet 2017;25:262–266.ArticlePubMedPMCPDF
REFERENCES
Figure & Data
References
Citations
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite