 E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 5(1); 2012 > Article
-
Case Report
Levodopa-Induced Facial Dystonia in a Case of Progressive Supranuclear Palsy - Eun Joo Chung, Sang Jin Kim
-
Journal of Movement Disorders 2012;5(1):28-32.
DOI: https://doi.org/10.14802/jmd.12008
Published online: April 30, 2012
Department of Neurology, Busan Paik Hospital, Inje University College of Medicine, Busan, Korea
- Corresponding author: Sang Jin Kim, MD, PhD Department of Neurology, Busan Paik Hospital, Inje University College of Medicine, 75 Bokji-ro, Busanjin-gu, Busan 614-735, Korea, Tel +82-51-890-6425, Fax +82-51-895-6367 E-mail jsk120@hanmail.net
- The authors have no financial conflicts of interest.
• Received: March 13, 2012 • Revised: April 23, 2012 • Accepted: April 23, 2012
Copyright © 2012 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 16,962 Views
- 67 Download
- 4 Crossref
ABSTRACT
- Progressive supranuclear palsy (PSP) is frequently misdiagnosed as other Parkinsonism because of clinical heterogeneity of PSP. We present here a case of a 67-year-old male patient with frontotemporal dementia-like cognitive impairment including language difficulties and abnormal behaviors. He showed severe facial dystonia after the levodopa treatment. Herein, we describe an unusual case of a patient presenting with PSP which, we believe could contribute to our knowledge about atypical leveodopa-induced facial dystonia in PSP.
- Clinical course
- A 67-year-old man was admitted to our hospital presenting gait disturbance and facial dystonia. His wife noted that he showed nonfluent speech having difficulty in naming objects and abnormal behavior such as hyperphagia for the first time 6 years previously. Thereafter, tremor in the lower limbs, more dominantly in the left leg, gait disturbance and postural instability insidiously progressed for 4 years. Since the age of 65, he has shown personality change and memory impairment. He had been known to be a very gentle and quiet person, but now often showed temperament. His memory worsened and cognitive function continued to decline. One year previously, he began to develop stooped posture and left shuffled gait which gradually worsened. By then, his movements slowed considerably, his gait became unsteady, and he fell frequently. A trial of levodopa (200 mg/ day) was prescribed dose subsequently increased to 600 mg per day over one year. However, no objective signs of improvement showed despite increased dose of levodopa, and involuntary facial dystonia began to develop.
- Neurological examination taken after the levodopa medication revealed severe facial dystonia. Involuntary facial grimacing usually sustained around both eyes accompanied with hypertonicity in facial muscles and deep facial folds. His speech was more difficult because of simultaneous contraction of facial muscles. Body bradykinesia, axial rigidity and rest tremor in both limbs, and rest tremor in both arms and legs, which was more severe on the left limbs, were shown. The motor score of the Unified Parkinson’s Disease Rating Scale (UPDRS) scored 59. Despite no definite supranuclear palsy, optokinetic nystagmus was not found. Motor power was intact and sensory examination was normal. There was no pathologic reflex on both feet despite increased overall deep tendon reflexes. The patient could not stand without assistance and fell down frequently.
- After the discontinuation of levodopa, motor score of UPDRS dropped to 38 points and facial dystonia completely disappeared. There was bilateral symmetric parkinsonism on the proximal parts. When answering questions or having conversation, he showed groping speeches, intermittent palilalia and severe anomia. However, single word comprehension was preserved. A month later, levodopa administration restarted and doses were gradually increased to 150 mg per day. This increased medication caused reemergence of his facial dystonia. Motor score of UPDRS worsened to 45 points.
- Results of routine laboratory investigations for dementia with Parkinsonism and gene (MAPT and progranulin) study for FTD were normal. Fourteen months later, at the age of 68 years, he became completely unable to walk even with assistance. His upward and downward gazes were absent and he finally showed supranuclear palsy.
- Neuropsychologic evaluation
- Results of neuropsychologic assessment performed 3 years previously showed naming difficulty (12.1 percentile) and severely impaired frontal executive function (2.68 percentile). Verbal and visual memory, attention, and visuospatial function were still preserved. Follow-up neuropsychologic assessment in our laboratory also showed a more significant decline in Boston naming test result (4.85 percentile) and frontal executive function (0.02 percentile) than the past examination. In addition, the performance of visuospatial function evaluated with the Rey Complex Copy test and memory ability evaluated with auditory verbal learning test marked several standard deviations below those of normal control subjects. Because of facial dystonia, he seldom spoke during hospitalization. He underwent the Korean version of the Western Aphasia Battery (KWAB) after the discontinuation of levodopa. Language was severely impaired, based on KWAB test results. Speech was non-fluent and marked by anomia. Comprehension was normal but repetition was impaired. Mini-Mental State Examination scored decreased to 26 from 29 in 2 years. His neuropsychologic results were summarized in Table 1.
- Neuroimaging studies
- Magnetic resonance images of the brain showed markedly bilateral frontotemporal lobe, midbrain and cereballar atrophy (Figure 1). 18F-fluorodeoxyglucose positron emission tomography showed hypometabolism definitely in the bilateral frontotemporal area (Figure 2). Lesser hypometabolic deficits were observed in the left parietal lobe. 123I-metaiodobenzylguanidine scintigraphy revealed normal early and delayed H/M ratios (early H/M ratio: 1.91, delayed H/M ratio: 2.38).
Case
- Language problem of the patient insidiously and slowly progressed once started. In addition, anomia and impaired repetition, which were documented by KWAB, were supportive of the diagnostic features of PNFA.2 Therefore, this initially led us to clinically suspect his condition as FTD. On the other hand, the MAPT and progranulin, the most popular genes in the FTD,13 were not found.
- Despite the lack of typical clinical symptoms of PSP in the early stage, this patient showed some positive features that conform to the criteria,1 which are symmetric proximal parkinsonism, retrocolic posture, poor response of levodopa, and early onset cognitive impairment including frontal dysfunction. Over the six years from the symptom onset, gait disturbance and falling gradually became more severe and extraocular movement impairment began to show (from initial negative optokinetic nystagmus to supranuclear palsy eventually). These findings led to the diagnosis of clinically probable PSP.
- Facial dystonia in this patient was thought to be a motor complication from levodopa therapy and could be considered as a form of levodopa-induced dyskinesia (LID). The development of LID in Parkinson’s disease usually depends on neuroplastic changes in the basal ganglia circuitry caused by degeneration of nigrostriatal dopaminergic neurons after the prolonged levodopa treatment.14 In contrast, the rarity of dyskinesias in PSP is associated with multilevel damage in the nigrostriatal and striatopallidal systems.15 Abnormal movements of this patient included facial dystonia, which occurred when the patient was on levodopa medication.
- In conclusion, we presented a PSP patient with FTD-like behavioral problems. Although LIFD was rarely reported in PSP, our case suggested that LIFD could be considered as an atypical feature of PSP.
Discussion
Figure 1.Brain MRI. MRI shows diffuse cerebral cortex (especially bilateral fr-onto-temporal lobe), brain stem and cerebellar atrophy. In particular, midbrain atrophy is shown.
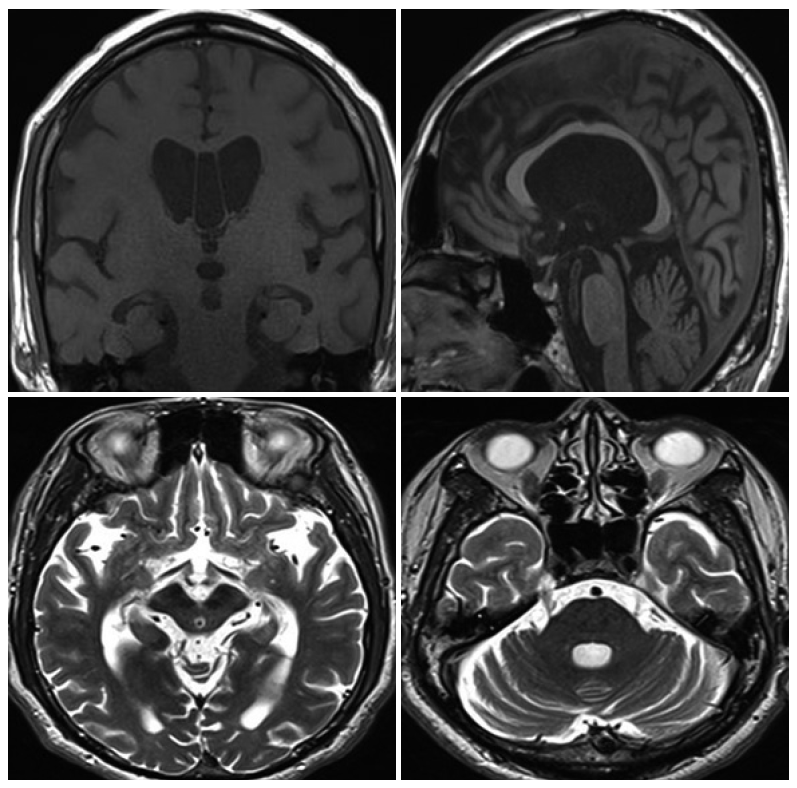
Figure 2.F-18 fluorodeoxyglucose positron emission tomography (FDG-PET) image. The FDG-PET showed decreased metabolism in caudate nucleus, bilateral frontal, temporal and parietal cortex, especially lower FDG uptake in the left hemisphere.
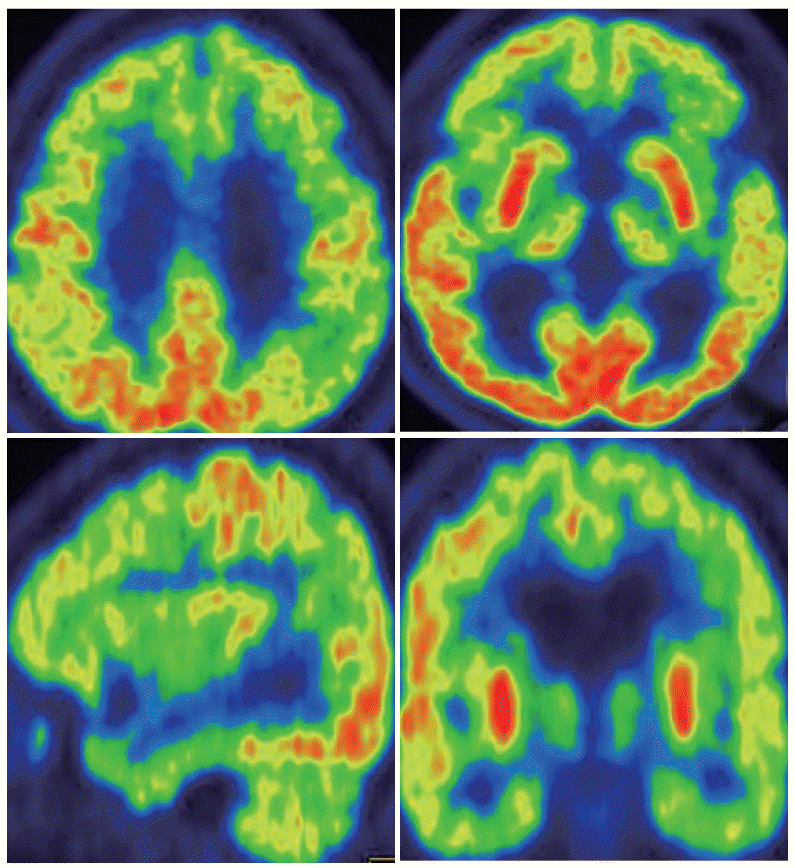
Table 1.Neuropsychological performances
- 1. Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology 1996;47:1–9.ArticlePubMed
- 2. Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998;51:1546–1554.ArticlePubMed
- 3. Mochizuki A, Ueda Y, Komatsuzaki Y, Tsuchiya K, Arai T, Shoji S. Progressive supranuclear palsy presenting with primary progressive aphasia--clinicopathological report of an autopsy case. Acta Neuropathol 2003;105:610–614.ArticlePubMed
- 4. Rohrer JD, Paviour D, Bronstein AM, O’Sullivan SS, Lees A, Warren JD. Progressive supranuclear palsy syndrome presenting as progressive nonfluent aphasia: a neuropsychological and neuroimaging analysis. Mov Disord 2010;25:179–188.ArticlePubMedPMC
- 5. Boeve B, Dickson D, Duffy J, Bartleson J, Trenerry M, Petersen R. Progressive nonfluent aphasia and subsequent aphasic dementia associated with atypical progressive supranuclear palsy pathology. Eur Neurol 2003;49:72–78.ArticlePubMed
- 6. Josephs KA, Boeve BF, Duffy JR, Smith GE, Knopman DS, Parisi JE, et al. Atypical progressive supranuclear palsy underlying progressive apraxia of speech and nonfluent aphasia. Neurocase 2005;11:283–296.ArticlePubMed
- 7. Josephs KA, Duffy JR, Strand EA, Whitwell JL, Layton KF, Parisi JE, et al. Clinicopathological and imaging correlates of progressive aphasia and apraxia of speech. Brain 2006;129(Pt 6):1385–1398.ArticlePubMedPMC
- 8. Josephs KA, Petersen RC, Knopman DS, Boeve BF, Whitwell JL, Duffy JR. Clinicopathologic analysis of frontotemporal and cortico-basal degenerations and PSP. Neurology 2006;66:41–48.ArticlePubMed
- 9. Forman MS, Farmer J, Johnson JK, Clark CM, Arnold SE, Coslett HB, et al. Frontotemporal dementia: clinicopathological correlations. Ann Neurol 2006;59:952–962.ArticlePubMedPMC
- 10. Josephs KA, Duffy JR. Apraxia of speech and nonfluent aphasia: a new clinical marker for corticobasal degeneration and progressive supranuclear palsy. Curr Opin Neurol 2008;21:688–692.ArticlePubMed
- 11. Barclay CL, Lang AE. Dystonia in progressive supranuclear palsy. J Neurol Neurosurg Psychiatry 1997;62:352–356.ArticlePubMedPMC
- 12. Tan EK, Chan LL, Wong MC. Levodopa-induced oromandibular dystonia in progressive supranuclear palsy. Clin Neurol Neurosurg 2003;105:132–134.ArticlePubMed
- 13. Rabinovici GD, Miller BL. Frontotemporal lobar degeneration: epidemiology, pathophysiology, diagnosis and management. CNS Drugs 2010;24:375–398.ArticlePubMedPMC
- 14. Olanow CW, Agid Y, Mizuno Y, Albanese A, Bonuccelli U, Damier P, et al. Levodopa in the treatment of Parkinson’s disease: current controversies. Mov Disord 2004;19:997–1005.ArticlePubMed
- 15. Kim JM, Lee KH, Choi YL, Choe GY, Jeon BS. Levodopa-induced dyskinesia in an autopsy-proven case of progressive supranuclear palsy. Mov Disord 2002;17:1089–1090.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
Citations to this article as recorded by 
- Use of botulinum toxin in the management of dystonia in Parkinson’s disease
Charenya Anandan, Joseph Jankovic
Frontiers in Neuroscience.2024;[Epub] CrossRef - Lower Cranial Dystonia with Inflated Cheeks: A Case of Dystonic Respiratory Failure
Takashi Suzuki, Takao Makifuchi, Nobuyoshi Fukuhara
Internal Medicine.2023; 62(11): 1671. CrossRef - Dystonia in atypical parkinsonian disorders
Luca Marsili, Matteo Bologna, Maja Kojovic, Alfredo Berardelli, Alberto J. Espay, Carlo Colosimo
Parkinsonism & Related Disorders.2019; 66: 25. CrossRef - A Review of Treatment Options for Progressive Supranuclear Palsy
Maria Stamelou, Günter Höglinger
CNS Drugs.2016; 30(7): 629. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite

