E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 2(1); 2009 > Article
-
Case Report
Adult Onset Familial Cherry-Red Spot Myoclonus - Chi Kyung Kima, Beom S. Jeonb
-
Journal of Movement Disorders 2009;2(1):50-52.
DOI: https://doi.org/10.14802/jmd.09014
Published online: April 30, 2009
aMovement Disorder Center, Seoul National University Hospital, Seoul, Korea
bDepartment of Neurology, Seoul National University Hospital, Seoul, Korea
- Corresponding author: Beom S. Jeon, MD, PhD, Department of Neurology, Seoul National University Hospital, 101 Daehak-ro, Jongno-gu, Seoul 110-744, Korea, Tel +82-2-2072-2876, Fax +82-2-2072-0839, E-mail brain@snu.ac.kr
• Received: March 20, 2009 • Accepted: March 23, 2009
Copyright © 2009 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 18,688 Views
- 55 Download
ABSTRACT
- We report a case of a 36-year-old woman with progressive generalized myoclonus that first became apparent 9 years ago. Her younger brother had similar problems. Examination of her eyes revealed cherry-red spots. Hexosaminidase A, β-galactosidase and neuraminidase activity were normal. Although the laboratory findings were negative, cherry-red spots, progressive myoclonus and autosomal recessive inheritance pattern suggested that she had an unknown type of lysosomal storage disease.
- A 36-year-old woman complained of an insidious onset of generalized myoclonus that first became apparent at age 27 years. She had no perinatal problems and her development was normal in childhood and juvenile periods. Adult-onset myoclonus had worsened progressively from right hand to four extremities, tremulous voice and gait disturbance developed after 3 years from disease onset, and she could not continue working as a nurse. She did not complain decreased visual acuity and hearing difficulty. She had no history of febrile convulsions or seizure, infectious disease in the central nervous system, exposure to toxic materials, or intake of herbal drugs.
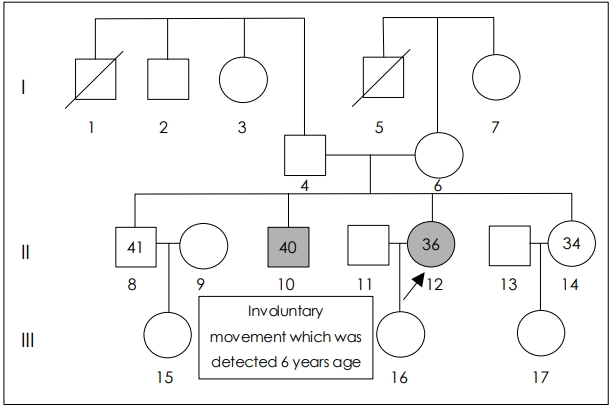
- Her younger brother aged 40 years old also had progressive generalized myoclonus, which was detected 6 years ago at age 34 years (Figure 1). He had normal intelligence and did not complain decreased visual acuity. Other family members except younger brother were reportedly healthy.
- A physical examination did not reveal any dysmorphism or evidence of hepatomegaly. The patient was alert and oriented, and her Mini-Mental State Examination score was 30. She did not have gaze palsy, and her vision and hearing were normal; however, her voice was tremulous and generalized positive myoclonus was observed at four extremities and body. Negative myoclonus, dystonia, tremor, and rigidity were not detected, and both motor and sensory functions were intact. Deep tendon reflexes were normal, and Babinski’s sign was not present. There was no evidence of cerebellar dysfunction, and she did not have an ataxic or parkinsonian gait, although she staggered slightly because of myoclonus.
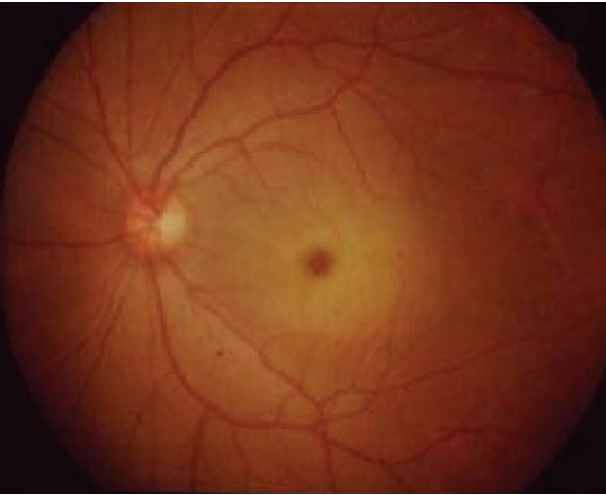
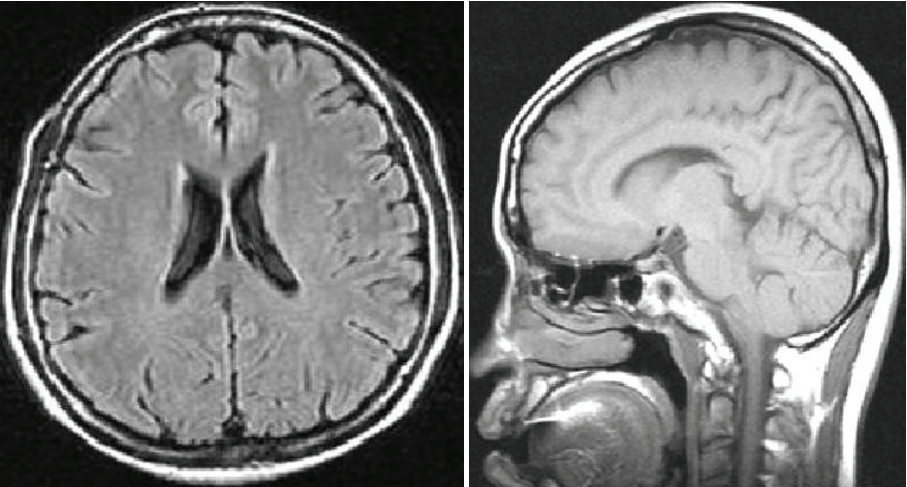
- Examination of the eyes revealed cherry-red spots (Figure 2), but her electroencephalogram was normal. Levels of serum electolytes, creatinine, and liver enzymes were normal. No white matter lesion or cerebellar atrophy was detected in an MRI of her brain (Figure 3).
- Neuraminidase, hexosaminidase A, and β-galactosidase activities in the leukocytes and cultured fibroblasts in patient and younger brother were normal.
Case Report
- The cherry-red spot is a pale perifoveal ring that develops when large deposits of lipid, sphingolipid, or oligosaccharide material accumulate in the ganglionic cells at the macula.2 This is a characteristic finding in storage diseases, including the sialidoses, GM1 and GM2 gangliosidoses, neuronal ceroid lipofuscinosis, Niemann-Pick disease (groups A through D), Farber’s lipogranulomatosis, and metachromatic leukodystrophy. Interestingly, Niemann-Pick disease, Farber’s lipogranulomatosis, and metachromatic leukodystrophy are not associated with myoclonus. Moreover, the patient in this report did not have typical findings of these 3 diseases such as the organomegaly, cognitive impairment, and gaze palsy seen in Niemann-Pick disease4; the hoarseness, arthritis, and subcutaneous nodules seen in Farber’s lipogranulomatosis; or the abnormal brain MRI findings in metachromatic leukodystrophy.
- In ceroid lipofuscinosis, sialidosis, GM1 and GM2 gangliosidoses, myoclonus, and maculopathy (e.g. a cherry-red spot) may coexist. Although they are quite similar in appearance, the macular abnormality seen in patients with neuronal ceroid lipofuscinosis (which has been described as “bull’s-eye maculopathy”) can be distinguished from the cherry-red spot by color and shape, as well as by the decreased visual acuity and visual-field restriction that are common in neuronal ceroid lipofuscinosis.5 Moreover, adult-onset lipofuscinosis has an autosomal dominant inheritance, rather than the autosomal recessive pattern seen in this patient.6
- GM1 gangliosidosis results from a deficiency of β-galatosidase; the adult form (type 3) presents as a slowly progressive dementia with prominent parkinsonian features and extra-pyramidal dysfunction, particularly dystonia.7 GM2 gangliosidosis results from a deficiency of hexosaminidase A; the late form (with an onset during adolescence and young adulthood) may be characterized by cognitive dysfunction, cerebellar dysfunction, upper and lower motor neuron involvement, and extrapyramidal dysfunction.8 Because β-galatosidase and hexosaminidase activity was normal in this patient, because she has a normal level of intelligence, and because no other prominent pyramidal or extrapyramidal dysfunction was detected, we might not diagnose this patient as GM1 and GM2 gangliosidoses.
- Sialidosis is an inherited, autosomal recessive disease associated with a neuraminidase deficiency.9 It has 2 major clinical manifestations: type I (late, adult onset) and type II (early, infantile onset). Type I sialidosis is typically found in patients aged 8 to 25 years and is characterized by cherry-red spot myoclonus, seizure, neuropathy, corneal clouding, and difficulty walking, but with normal vision and intelligence (O’Brien, 1978). Type II sialidosis is characterized by dysmorphism, myoclonus, mental retardation, ocular cherry-red spots, and hepatosplenomegaly.
- The patient in this report may present clinical evidence of type I sialidosis, but her laboratory findings do not support this diagnosis; activities of the neuraminidase were normal. A similar case of progressive myoclonic epilepsy has been reported.10 Differences from the previous report which described a patient with progressive myoclonic epilepsy, cherry-red spots and negative enzyme deficiency were theses; 1) the onset-age was older than the previous report (27 versus 13 years, adult versus juvenile-onset); 2) the patient had a sibling with same disease, which indicated she had inherited disease, but the patient in previous report did not have familial history; 3) the patient in our report did not have a history of seizure, and it is different from the patient with myoclonic epilepsy.
- Although the cause of cherry-red spot myoclonus is not clear, to our knowledge, this is the first report of adult-onset familial cherry-red spot myoclonus caused by an unknown type of lysosomal storage disease in Korea.
Discussion
Figure 1.Pedigree of the patient. Arrow indicates the patient. Gray color indicates an affected person. Square box indicates male and circle indicates female. I-1 deceased of old age, and I-5 deceased in the war.

- 1. Caviness JN, Brown P. Myoclonus: current concepts and recent advances. Lancet Neurol 2004;3:598–607.ArticlePubMed
- 2. Leavitt JA, Kotagal S. The “cherry red” spot. Pediatr Neurol 2007;37:74–75.ArticlePubMed
- 3. Danon MJ, Oh SJ, DiMauro S, Manaligod JR, Eastwood A, Naidu S, et al. Lysosomal glycogen storage disease with normal acid maltase. Neurology 1981;31:51–57.ArticlePubMed
- 4. Kolodny EH. Niemann-Pick disease. Curr Opin Hematol 2000;7:48–52.ArticlePubMed
- 5. Collins J, Holder GE, Herbert H, Adams GG. Batten disease: features to facilitate early diagnosis. Br J Ophthalmol 2006;90:1119–1124.ArticlePubMedPMC
- 6. Burneo JG, Arnold T, Palmer CA, Kuzniecky RI, Oh SJ, Faught E. Adult-onset neuronal ceroid lipofuscinosis (Kufs disease) with autosomal dominant inheritance in Alabama. Epilepsia 2003;44:841–846.ArticlePubMed
- 7. Muthane U, Chickabasaviah Y, Kaneski C, Shankar SK, Narayanappa G, Christopher R, et al. Clinical features of adult GM1 gangliosidosis: report of three Indian patients and review of 40 cases. Mov Disord 2004;19:1334–1341.ArticlePubMed
- 8. Frey LC, Ringel SP, Filley CM. The natural history of cognitive dysfunction in late-onset GM2 gangliosidosis. Arch Neurol 2005;62:989–994.ArticlePubMed
- 9. Lowden JA, O’Brien JS. Sialidosis: a review of human neuraminidase deficiency. Am J Hum Genet 1979;31:1–18.PubMedPMC
- 10. Kang KS, Yun CH, Lee SK. Progressive Myoclonus Epilepsy Associated with Macular Cherry-Red Spots. J Korean Neurol Assoc 2003;21:204–206.
REFERENCES
Figure & Data
References
Citations
Citations to this article as recorded by 
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite