Treatment of Gait Ignition Failure with Ropinirole
Article information
Abstract
Gait ignition failure (GIF) is a syndrome characterized by hesitation or inability to initiate gait from a static position. It may occur in a variety of conditions, including normal pressure hydrocephalus, subcortical vascular disease, parkinsonian syndromes and a variety of focal lesions. Previous information on the treatment of GIF has been primarily anecdotal, but there have been a few reports of response to dopamine agonists. We report a 63-year-old man with anoxic encephalopathy who developed GIF nine years after the initial anoxic insult. The patient’s GIF responded robustly, albeit transiently, to ropinirole. MRI was unrevealing, but a positron emission tomography scan showed hypometabolism in the deep frontal ACA/MCA watershed area; this may have disconnected the basal ganglia from the motor cortex and/or interrupted dopaminergic mesocortical transmission. Our understanding of the pathophysiology and the treatment of GIF remains limited, but there may be at least a limited therapeutic role for dopamine agonists.
Gait ignition failure (GIF) is a syndrome characterized by a hesitation or inability to initiate gait from a static position [1,2]. Patients appear as if their feet are stuck to the floor or march in place with stuttering, non-propulsive steps [3]; once this hesitation is overcome, gait is normal, or at least in the patient’s normal pattern. Gait will often arrest again during turns, while traversing narrow spaces such as doorways, or when distracted. GIF has many names, including magnetic apraxia, apractic gait, slipping-clutch gait and Brun’s ataxia [1].
Gait ignition failure is observed in a variety of neurologic illnesses, including normal pressure hydrocephalus, subcortical vascular disease, many parkinsonian syndromes [idiopathic Parkinson’s disease (PD), progressive supranuclear palsy, corticobasal degeneration, multiple systems atrophy] and a variety of focal lesions [1,2]. Factor et al. [1] have suggested that GIF may occur in isolation as a discrete neurologic entity (“Primary Progressive Freezing Gait”), although all of the patients in their series ultimately developed prominent parkinsonian features.
Previous information on the treatment of GIF has been primarily anecdotal. It has been suggested that patients may be able to circumvent the problem by using strategies that recruit the pyramidal system and supplementary motor area, such as trying to step on specific spots on the ground, imagining an obstacle to step over or using music for pacing [4], but this been studied mostly in PD. Pharmacologic treatment has mostly focused on dopaminergic agents, most likely because of the occurrence of the syndrome in various parkinsonian illnesses. Levodopa has invariably been reported as ineffective except in advanced PD [2,3], whereas Taskapilioglu et al. [2] have reported several patients whose GIF responded to dopamine agonists. We recently cared for a patient whose GIF responded quite well, albeit transiently, to treatment with ropinirole.
CASE REPORT
A 63-year-old man, a nursing home resident for ten years, had been admitted six months after an episode of anoxic encephalopathy. The sequelae included severe memory and executive impairment with anosognosia and irritability, as well as visual impairment consistent with apperceptive agnosia. Past medical history was significant for traumatic T10-L1 compression fractures approximately 35 years prior to the anoxic event, resulting in paraparesis which gradually resolved with laminectomy/fusion and physical therapy; we have no information about his gait prior to the anoxic episode although he had been functionally independent. Past history was also significant for hyperlipidemia and gastroesophageal reflux disease. He had been on neuroleptics since admission due to irritability and volatility, initially olanzapine 5 mg qhs for approximately four years and subsequently risperidone in doses between 1 mg and 2 mg bid. Other medications included baclofen 15 mg bid, citalopram, simvastatin and omeprazole.
At baseline, his gait was fast and impulsive, hypermetric with a wide base, strong heel strike and arms held in abduction; he fell frequently, mostly as a result of his poor safety awareness and visual impairment. He was unaware of his gait problems and was always curiously reticent to use a wheelchair. There had never been any problems with gait initiation nor any signs of parkinsonism.
Approximately nine years after the anoxic insult, he began to develop problems with gait initiation, characterized by taking multiple stuttering in-place steps before starting and to a lesser extent in association with turns. This gradually worsened from lasting only a few seconds to 30 or more seconds. As the GIF worsened, he was noted to be unable to shift his center of gravity forward, and he bore his weight on his heels; he ultimately fell backward a few times while trying to start. Once he began walking, his gait was otherwise unchanged from the baseline pattern. There were no other new neurologic signs or symptoms, no signs of parkinsonism and no loss of postural reflexes. There were no recent illnesses or medication changes. Anatomical scanning by MRI was unchanged from a scan six years previously, showing old areas of bilateral posterior temporal and occipital encephalomalacia. Functional imaging by 18F-fluorodeoxyglucose positron emission tomography (PET) scan (Figure 1) showed hypometabolism in these same areas and in the bilateral deep frontal anterior cerebral artery/middle cerebral artery watershed area as well as bilateral striatal hypermetabolism.
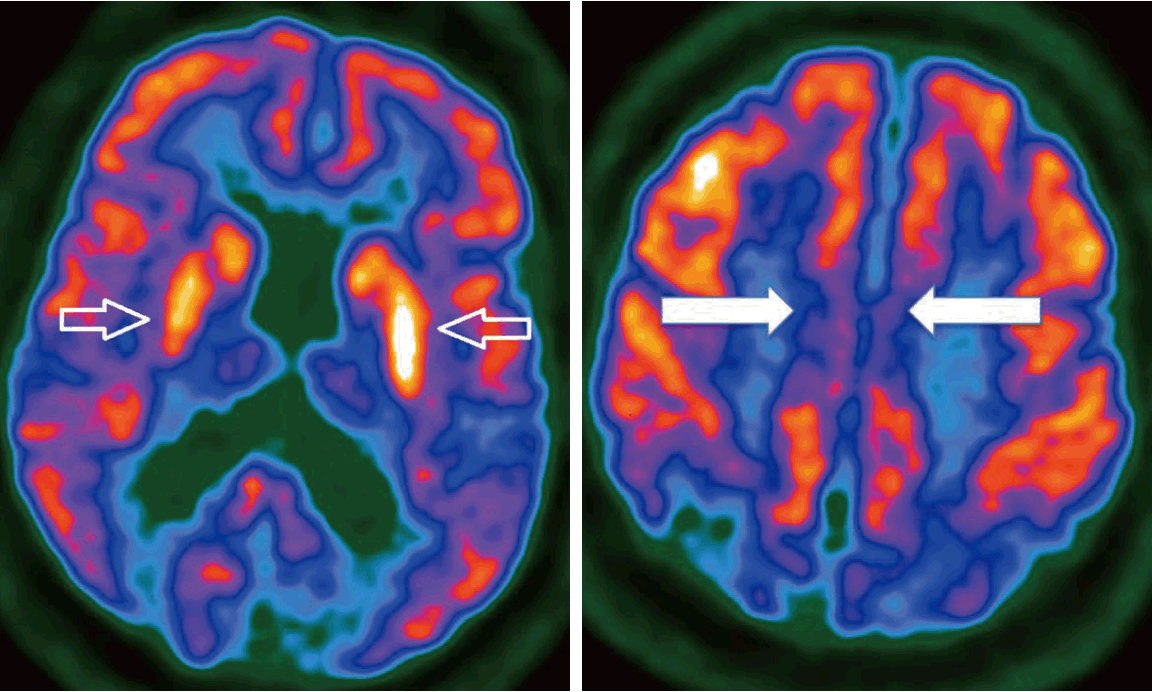
Axial positron emission tomography scan showing areas of hypometabolism in the deep ACA/MCA watershed area (solid arrows) and hypermetabolism in the striatum (hollow arrows).
We initially changed the neuroleptic from risperidone to olanzapine (to which he had previously responded) 5 mg qhs to reduce any possible extrapyramidal burden; this was ineffective, and his behavior deteriorated, necessitating restarting risperidone after 17 days at a lower dosage of 1 mg bid. Kinesiotherapy found that he was unable to see visual pacing cues on the floor nor to use imagery; audible cuing with a metronome was ineffective. Passively moving his center of gravity forward to initiate a step was effective but did not generalize, and there was no obvious benefit to practicing starting and stopping on a treadmill.
After four months we began a trial of ropinirole, titrating over a period of four weeks to a maximum dosage of 4 mg/d. This resulted in a robust improvement in the GIF, with usually no stuttering steps and never any more than two or three on gait initiation. He tolerated ropinirole with no untoward effects. After approximately two months he began refusing the ropinirole. The GIF returned, with anywhere from eight to 20 in-place steps before gait initiation and on turning. After approximately two weeks, we convinced him to restart ropinirole; this was again titrated to a dosage of 6 mg/d, and the GIF again improved as previously. He continued to do well with near-complete resolution of the GIF for the next three months, at which time the problem gradually recurred. He again began to refuse the ropinirole, and we discontinued further trials.
DISCUSSION
Although the improvement in our patient’s GIF may have been attributable to kinesiotherapy, its recurrence on discontinuing the ropinirole and subsequent three-month improvement on restarting the drug was essentially an A-B-A design and is highly suggestive of a pharmacologic response. Likewise, the GIF appeared clearly independent of the changes in neuroleptics. Quetiapine or clozapine would have been a better choice to more completely rule out the role of neuroleptics in the GIF, but he had previously responded well to olanzapine, and olanzapine is associated with less extrapyramidal burden than risperidone [5]. Overall, it appears unlikely that the neuroleptics had contributed to the GIF, as he had been on a stable dose of risperidone for approximately five years, as he had no other extrapyramidal signs, and as GIF is rare in drug-induced parkinsonism.
Although many previous reports of GIF have been associated with parkinsonian syndromes [1,2], our patient has not developed any other parkinsonian signs after one year of follow-up, and we suspect that the GIF is causally related to his anoxic brain damage.
There have been several reports of GIF, absent other parkinsonian signs, following anoxic brain damage [3,6]. Pathophysiologically, the basal ganglia are particularly sensitive to hypoxic damage, and several authors have implicated these areas in GIF [3]. The anterior watershed area is also especially sensitive to hypoxia and has also often been implicated in the pathogenesis of GIF [2,6]. The striatal hypermetabolism on our patient’s PET scan may in fact be related to loss of reciprocal inhibition by the frontal cortex, which was undercut by the damaged ACA/ MCA watershed area. Several authors have suggested that GIF can result when the supplementary motor area and motor cortex are thus disconnected from the basal ganglia [2,3,7].
It is not known whether GIF is necessarily related to a dopaminergic deficit. Rascol et al. [8] have suggested that diseases of dopaminergic systems can cause GIF and that in some patients with PD, GIF can be observed in the “off ” periods and can respond to levodopa. If the anoxic injury to the deep frontal lobes indeed resulted in a disruption of dopaminergic transmission between the midbrain and the frontal lobes, then this might explain the efficacy of a dopamine agonist in our patient.
Although it is conceivable that our patient’s GIF may be related to an as-yet unmanifested parkinsonian syndrome, we suspect that it is causally related to the anoxic insult nine years previously. This delayed onset has been noted in other cases of anoxiainduced GIF [6]. Satz [9] has discussed the concept of brain reserve capacity, observing that aging or other insults can deplete this capacity and that this can lead to an earlier onset of dementia and other neurologic problems as the brain ages. This model has been especially prominent in explaining the increased incidence of dementia in patients with a history of traumatic brain injury [10]. It appears reasonable, to speculate that this model may pertain to high-order gait changes as much as to cognitive changes and to anoxic damage as much as to mechanical damage. We suspect that our patient suffered anoxic damage to the watershed areas and that the resultant GIF has manifested only after nine years of age-related loss of reserve. This model of progressive loss of reserve might also explain the ultimate loss of efficacy of the ropinirole.
Gait ignition failure is a tremendously disabling syndrome and is most likely associated with high fall risk. Our understanding of the pathophysiology and the treatment of GIF remains limited, but there may be at least a limited therapeutic role of dopamine agonists in its management.
Notes
Conflicts of Interest
The authors have no financial conflicts of interest.
Acknowledgements
This work was presented at the Neuroscience Chair Summit, Tampa, Florida, USA, September 2013.