Creutzfeldt-Jakob Disease in a Tertiary Care Hospital in Thailand: A Case Series and Review of the Literature
Article information
Abstract
Creutzfeldt-Jakob Disease (CJD) is an incurable and inevitably fatal neurodegenerative disorder. Although CJD has a worldwide distribution, there are no official statistics on CJD in Thailand. A diagnosis of CJD is suspected when a patient develops rapidly progressive dementia with myoclonus. However, CJD may be mistaken for a variety of illnesses because its initial presentation frequently consists of non-specific symptoms. Here, we examined cases of sporadic CJD (sCJD) from Thammasat University Hospital (a tertiary care hospital in Thailand) between January 1, 2012 and December 31, 2014. Three cases of probable and possible sCJD were collected. All cases presented with rapidly progressive cognitive dysfunction accompanied by spontaneous myoclonus. Classical electroencehalography changes and typical abnormal MRI features were observed. All of the cases died within a period of 8 months. None of the patients underwent brain biopsy. Our findings raise questions about the prevalence of CJD in Thailand, which needs further study.
Creutzfeldt-Jakob Disease (CJD) is a rare, incurable, and inevitably fatal neurodegenerative disorder. CJD is the most common of the known human transmissible spongiform encephalopathies that are caused by abnormal folding of specific cellular proteins called Prion Proteins. The accumulation of these abnormal proteins disrupts cell function and causes a sponge-like appearance in brain tissue. CJD has a worldwide distribution with an annual incidence of between 0.5–1 cases per million individuals in the general population. In Thailand, there are no official statistics on or national surveillance of CJD. It was estimated that no more than 30 cases have been diagnosed in the previous 20 years. Here, we report two cases of patients with probable sporadic CJD (sCJD) and one case with possible sCJD from Thammasat University Hospital between January 1, 2012 and December 31, 2014 (Table 1).

Clinical presentations and investigations of cases
CASE REPORT
Case 1
A 42-year-old Thai woman presented with sub-acute dizziness and an unsteady gait. Two weeks later, she developed memory impairment and deterioration of concentration. Her interest and pleasure in all activities, as well as her speech, diminished remarkably. Her condition became progressively worse, and eventually she became akinetic-rigid and mute. Later, spontaneous myoclonic jerks were present in her distal limbs. Magnetic resonance imaging (MRI) showed high signal intensity in the bilateral caudate nucleus, putamen and left frontotemporal cortex on T2-weighted and diffusion-weighted imaging (DWI). Electroencephalography (EEG) revealed generalized and synchronous periodic sharp wave complexes occurring at intervals of 0.6–0.8 seconds. A back-averaging EEG confirmed cortical origin multifocal myoclonus. She died 32 weeks after the initial presentation. Her clinical history together with physical signs, typical EEG, and brain MRI findings were consistent with a diagnosis of probable sCJD [1].
Case 2
A 76-year-old Thai man presented with a three-week history of confusion, hallucinations, dysarthria, and mild left-sided weakness. He denied a history of headache or fever. His general blood chemistry and cerebrospinal fluid (CSF) studies were within a normal range. A brain MRI study showed symmetric hypersignal intensity on fluid-attenuated inversion recovery (FLAIR) MRI sequences with restricted diffusion on DWI in his bilateral caudate nuclei, putamen and bilateral fronto-temporo-parietal cortices. An EEG revealed periodic generalized sharp wave complexes. His clinical status gradually deteriorated with time, and he developed spontaneous generalized myoclonic jerks. He developed aspiration pneumonia and passed away, succumbing to the illness 8 months after its onset. The patient was diagnosed as having probable sCJD [1].
Case 3
A 53-year-old Thai female teacher presented with a history of dizziness, fatigue, and insomnia. One month later, she developed social withdrawal, psychomotor retardation, and memory impairment. On physical examination, she was alert but had markedly decreased speech output. Generalized rigidity and occasional spontaneous myoclonic jerks were observed. A tentative diagnosis of akinetic-rigid syndrome was made, and full blood examinations and CSF investigations were performed. A computed tomography (CT) brain scan was unremarkable. An EEG revealed an intermittent delta slow wave superimposed on a normal alpha background. Her symptoms rapidly deteriorated, and she became bedridden and mute. Multiple spontaneous limb myoclonic jerks were observed. She died 4 months after symptom onset. Her clinical history, progression and physical findings suggested a diagnosis of possible sCJD [1].
DISCUSSION
CJD is a human prion disease with characteristic clinical and diagnostic features. This fatal neurological disease occurs in sporadic, familial and acquired forms. sCJD is the most common type of CJD, accounting for at least 85 percent of cases [2]. In Thailand, there is limited literature on CJD, as it has been underreported and misdiagnosed. There is no regional or national surveillance system for CJD in Southeast Asia [3]. Furthermore, knowledge of this rare disease is limited to medical specialists. These facts reflect the need for public awareness, an effective reporting system, advanced medical education, and laboratory, neurological and neuropathological diagnostic capacity in this region. A literature search on the epidemiology of CJD in Southeast Asia revealed less than 25 published cases since the year 2000. There were no reports of variant, iatrogenic or familial CJD in this region [4,5]. CJD may be mistaken for a variety of illnesses because it typically initially presents as non-specific symptoms, such as dizziness, vertigo, insomnia, and personality and mood changes.
CJD has a degree of clinicopathological heterogeneity. Recently, six different molecular subtypes of sCJD have been identified, which vary with respect to age of onset, disease duration, presenting symptoms, neuropathology, and MRI signal alterations [6]. In general, the average age of onset and length of illness in patients with sCJD are 60 years and 4 months, respectively [2]. However, younger patients trend toward having prolonged survival times [7]. Cerebellar and vestibular features are common and can be the only presenting symptoms, especially in young patients, resulting in misdiagnosis [7,8]. Cognitive decline and myoclonus may be delayed for weeks or even months after disease onset, causing difficulty in diagnosis.
CJD can present as a movement disorder, and myoclonus is the most common abnormal movement that could occur at any stage; it can even be an initial manifestation of the illness. Myoclonic jerks are often generalized, asymmetric, cause greater effects to the distal extremities, and are associated with periodic sharp wave EEG activity [9]. Other movement disorders, including dystonia, choreoathetosis, tremor, hemiballismus, and atypical parkinsonian syndromes, have been described in a number of patients; however, these movements are usually combined with myoclonus [9]. Choreoathetosis is rare in sCJD but has been frequently reported in variant CJD (vCJD) and is listed in the diagnostic criteria for possible vCJD [10]. Atypical parkinsonian features, such as gait disorders, alien limb, and supranuclear gaze palsy, may occur as an initial presentation of CJD. Nevertheless, subacute progression rapidly combined with cognitive decline and myoclonus is suggestive of CJD [9]. Akinetic mutism (AM) is characterized by a marked reduction in motor function, including facial expression, gesture, and speech output, with preserved awareness. AM usually occurs in later stages of the disease and is considered a symptom that helps to establish the diagnosis of sCJD [1].
A definite diagnosis of CJD requires brain tissue examination, which is not performed routinely by many institutions due to the transmissible nature of the disease. Therefore, diagnostic tools such as EEG and assays of certain brain proteins in CSF have been used to define probable cases of CJD [3]. Recent studies using MRI have shown its potential as a diagnostic tool in sCJD, showing a sensitivity and specificity of 98% and 94%, respectively [1]. These changes comprise alterations on DWI and FLAIR MRI sequences in the caudate nucleus and putamen or in at least two cortical regions (temporal, parietal, and occipital) (Fig. 1A and B) [1]. An abnormality on DWI-MRI could be the first diagnostic clue of CJD and could be detected as early as 3 weeks after symptom initiation and even before the appearance of an abnormal EEG [11]. MRI can also be used to differentiate sCJD from vCJD. In vCJD, abnormalities are commonly located in the posterior and medial thalami followed by the periaqueductal grey matter, striatum, and less commonly the neo-cortex. An increased signal intensity in the posterior nuclei of the thalamus is known as a pulvinar sign and is the most sensitive marker for vCJD [11].
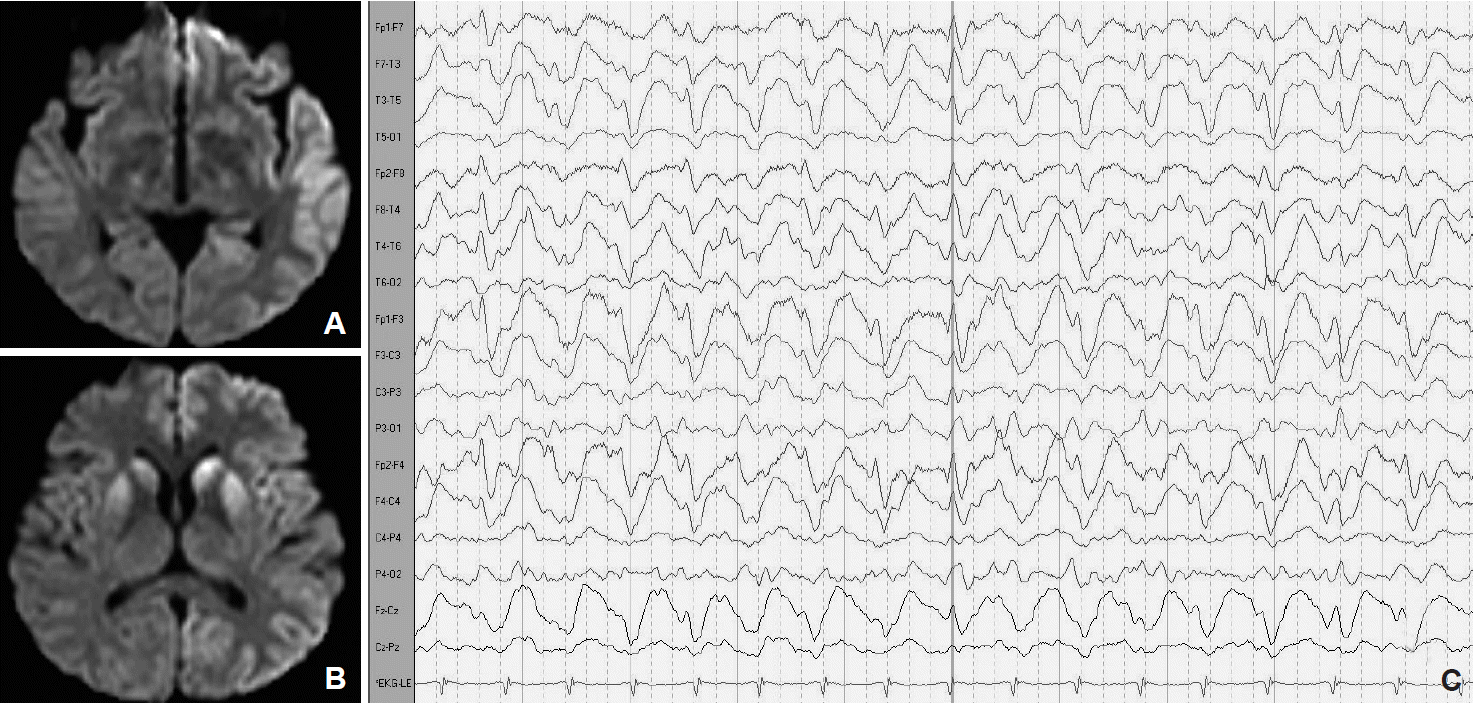
Diffusion-weighted magnetic resonance imaging showing increased signal intensity in the left frontotemporal cortex (A) and bilateral caudate nucleus and putamen (B). Periodic generalized sharp wave complexes detected by electroencehalography (C).
Several studies have demonstrated the benefit of evaluating 14-3-3 proteins in CSF for the diagnosis of sCJD [12]. However, an elevation of 14-3-3 proteins is not sensitive and specific for CJD. A number of other conditions that cause extensive neuronal damage, such as stroke, subarachnoid hemorrhage, hypoxic brain damage, encephalitis, and paraneoplastic encephalopathy, may also give positive 14-3-3 CSF results [3]. Another method is the use of a ratio of CSF total tau versus phosphorylated tau, which raised diagnostic specificity by up to 99%, but sensitivity remained relatively low at 79% [13]. Recently, the use of real-time quaking-induced conversion analysis of nasal brushings from olfactory epithelium has demonstrated a sensitivity of 97% and a specificity of 100% for the detection of CJD [14]. Unfortunately, these protein analysis assays are not available in Thailand.
EEG is an important diagnostic tool for the diagnosis of CJD. Approximately 60–80% of cases are reported to develop a characteristic appearance of 0.5–2 Hz periodic, generalized bursts of spike wave complexes (Fig. 1C). Typical EEG findings may be absent during the initial stages but often become apparent as the disease progresses. In most cases, abnormal EEG findings occur approximately 12 weeks after clinical onset. In our case series, characteristic EEG findings were found in 2 cases at 6 weeks after clinical onset, and they persisted to appear as abnormal at the end stage of the disease.
In conclusions, CJD is clinically heterogeneous and has a wide range of presenting symptoms. We report 3 cases with probable and possible sCJD presenting with non-specific clinical features and rapidly developed dementia, personality changes, parkinsonism culminating in AM and myoclonus during a 3-year period. EEG and MRI are important diagnostic tools that can detect abnormalities in early stages of the disease. Our findings raise questions about the true prevalence of CJD in Thailand, which needs further research and national surveillance. A definite and accurate diagnosis is crucial for patients, patients’ families and healthcare providers for preventing transmission and also for community surveillance of this infectious and transmissible condition.
Notes
Conflicts of Interest
The authors have no financial conflicts of interest.
Acknowledgements
We thank the patients and their families for their cooperation in this report.