Woodhouse-Sakati Syndrome: Report of the First Tunisian Family with the C2orf37 Gene Mutation
Article information
Abstract
Woodhouse-Sakati syndrome (WSS) is an infrequent autosomal recessive condition characterized by progressive extrapyramidal signs, mental retardation, hypogonadism, alopecia, and diabetes mellitus. This syndrome belongs to a heterogeneous group of inherited neurodegenerative disorders characterized iron accumulation in the brain, and it is caused by mutations of the C2orf37 gene. We report the first Tunisian family with two affected sisters presenting with a phenotype suggestive of WSS. We examined the index patient presenting with movement disorders and mental retardation and then searched for similar cases in her family, which identified a sister with similar signs. We performed a genetic study that confirmed the diagnosis and revealed a c.436delC mutation of the C2orf37 gene. Therefore, WSS is an important consideration in patients presenting with movement disorders and intellectual disability. A high consanguinity contributes to the clustering of such rare autosomal recessive syndromes.
Woodhouse-Sakati syndrome (WSS) is an infrequent autosomal recessive multi-systemic disorder. This syndrome, which was first described in 1983 by Woodhouse and Sakati [1], is characterized by hypogonadism, diabetes mellitus, alopecia, mental retardation and deafness. The age at disease onset, appearance of specific symptoms and symptom severity differs significantly among individuals with this syndrome and even among affected members of the same family. Such variability in the phenotype may be responsible for a delayed diagnosis or even misdiagnosis in some cases. Approximately 75 affected individuals from approximately 30 families have been reported in the literature [2]. Most of these families originate from the Middle East [2,3], particularly Saudi Arabia. Some cases were also observed in Northern and Eastern Europe [4,5], Turkey [6], India [7], and Pakistan [2]. The gene C2orf37, which is responsible for WSS, was first described in 2008 by Alazami et al. [8]. It is located on chromosome 2q22.3-q35. Currently, nine mutations have been reported in the literature.
We report here the first Tunisian family with two affected sisters suggestive of WSS on clinical presentation. The diagnoses were confirmed by molecular genetic analyses.
CASE REPORT
Informed written consent for the study, as well as permission to obtain a pedigree, present photographs for publication and to perform a genetic study, was obtained from elders including the parents of the affected individuals.
The index case is a 25-year-old woman referred to our department for her movement disorders. She is the sixth child of healthy first cousin parents (Figure 1). Her older sister is similarly affected, whereas the other siblings are healthy. Pregnancy, delivery, and early psychomotor development were unremarkable. The first symptom appeared at the age of 6 years when she was at school. The teacher noted learning disabilities and memory problems suggestive of mental retardation. At the age of 14, her parents noticed a progressive appearance of abnormal posturing and twisting movements that began in the arms, which eventually became generalized, leading to head shaking, postural impairment and walking difficulties. The age of puberty was surpassed and secondary sexual characteristics did not appear with primary amenorrhea. At the age of 18, she developed polydipsia with polyuria, and diabetes mellitus was discovered. Over ten years of follow-up, she became severely disabled and wheelchair bound because of her worsening movement disorder.

Pedigree showing consanguineous parents and the affected sisters (A), and photographs of the index case. Note the dysmorphic face and alopecia (B and C). The index patient is marked by an arrow.
Physical examination on admission disclosed generalized dystonia involving the trunk and all four limbs. Her face was dysmorphic, which included a triangular shape with a large forehead, alopecia predominant in the fronto-temporal region of the scalp, sparse eyebrows and eyelashes, hypertelorism, prominent nasal root, large ears, precocious skin aging and partial edentulism (Figure 1). Mild mental retardation was noted, along with an Mini-Mental State Examination score of 23/30. Secondary sexual characteristics were absent with no facial, axillary or pubic hair, and minimal breast budding was present with marked vulvar hypoplasia.
Laboratory investigations revealed a low estradiol level (13 pg/mL; normal range: 21–251 pg/mL), normal luteinizing hormone level (8.09 mIU/L, normal range 1.8–11.78 mIU/L) and high follicle-stimulating hormone level (39.1 mIU/L, normal range 3.03–8.08 mIU/L). These laboratory data are suggestive of hypogonadotropic hypogonadism. Insulin growth factor 1, prolactin and thyroid hormone (free thyroxin, thyroid-stimulating hormone) were within the normal ranges. An abdominal ultrasound showed uterine hypoplasia, and MRI of the brain showed T2 hypointensities of the globus pallidi (Figure 2) without white matter changes. Her ophthalmologic examination was unremarkable, and a hearing assessment excluded deafness. An electrocardiogram and electromyography with a nerve conduction velocity study revealed normal findings.
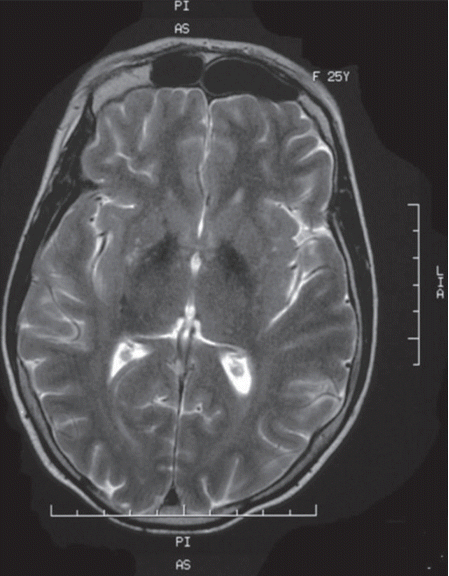
Brain MRI showing T2 hypointensities of the basal ganglia, particularly of the globus pallidi.
Her eldest sister, who was aged 30 years, presented at the age of 12 years with dystonic movement of the left upper limb. She worsened insidiously; the right side became affected 8 years later. At the age of 27 years, the dystonic movements spread to the lower limbs and resulted in walking difficulties with frequent falls. Her gait deteriorated progressively, and she became bedridden at the age of 28. She was mentally retarded, and she could not be integrated at school. She had a primary amenorrhea, and alopecia was noted since the age of 15 years. She demonstrated the same dysmorphic facial characteristics as her sister. She refused to be admitted to the hospital, and para-clinical investigation was limited to DNA sampling.
Blood was sampled from the two affected sisters, their parents and one healthy sister [IV: 5 on the pedigree (Figure 1)]. A genetic study was carried out in the Department of Neurology at King Faisal Specialist Hospital and Research Centre, Saudi Arabia. DNA was extracted from peripheral leukocytes according to standard procedures. All coding exons of the C2orf37 gene were amplified using polymerase chain reaction [8]. The primers were designed to flank the coding regions and exon/intron boundaries of C2orf37, as identified on the UCSC website (http://www.genome.ucsc.edu/), and were directly sequenced with the dideoxy chain-termination method [8]. Samples were processed on a Mega BACE 1000 (Molecular Dynamics, Sunnyvale, CA, USA), and the resultant chromatograms were assessed using the Seq Man II suite (DNASTAR, Madison, WI, USA) [8]. Sequencing analysis of DNA revealed the homozygous mutation c.436delC (p.Ala147Hisfs*9) in exon 4 of the C2orf37 gene in the index patient and confirmed the diagnosis of WSS. Further analyses revealed that this mutation was also present in a homozygous state in the other affected sister and in a heterozygous state in the parents and the healthy sister.
DISCUSSION
WSS is one of the inherited neurodegenerative disorders that are characterized by extrapyramidal movement disorders and abnormal iron accumulation in the basal ganglia of the brain [9]. The affected individuals in this family displayed the cardinal features of this syndrome: generalized dystonia, mental retardation, hypogonadotropic hypogonadism, alopecia and diabetes mellitus. No hearing loss was detected. Clinical diagnosis of WSS is based on the association of these characteristic manifestations; however, some of these symptoms may appear later, thus making the diagnosis very difficult in childhood. In fact, diabetes and extrapyramidal symptoms usually appear later in early adulthood. Additional manifestations, such as partial or complete edentulism, spastic quadriplegia and brain MRI abnormalities, are inconsistent features described in some cases. Partial edentulism, observed in our first case, has been reported in two other cases of WSS [2,4]. Brain MRI can reveal basal ganglia T2 hypointensities or white matter disease, which results from iron accumulation [9]. The clinical presentation in the two affected sisters in our Tunisian family is quite similar, but an intra familial phenotypic variability was previously noted by some authors [3].
The genetic study conducted by Alazami et al. [8] on 15 families contributed to the identification of the mutations of the C2orf37 gene that causes WSS. The mutation identified in our two cases is pathogenic and was previously described in person of Bedouin descent [3,8,10]. Despite the pronounced phenotypic variability of WSS, no correlation between phenotype and genotype has been proven. The pathogenesis of this multisystem disorder has been and still remains unclear. The C2orf37 gene is highly expressed in the brain, liver, and skin [8]. It was also shown that the nucleoli of lymphoblasts in individuals with WSS show enhanced sensitivity to transcriptional blockade [8], thus defective ribosome biogenesis and other nucleolar processes may be implicated in the pathogenic mechanisms of WSS [8]. Many hypotheses have been described in the literature, but further experimental studies are necessary to elucidate the exact pathogenic mechanisms of how these genes affect different tissues.
WSS should be considered in patients with movement disorders and mental retardation. Identification of a mutation in the C2orf37 gene is helpful for confirming the diagnosis of WSS, particularly in young patients in whom the characteristic symptoms of this syndrome have not yet manifested. Inbreeding and consanguineous marriages in the Arab world increase the risk of such rare autosomal recessive diseases among offspring. Based on the family presented here and other cases reported from this region, it seems that WSS is widely prevalent, mainly among Arabs, and is probably often misdiagnosed.
Notes
Conflicts of Interest
The authors have no financial conflicts of interest.
Acknowledgements
The authors thank all family numbers for their participation in this study.
This study was performed using the research funding of our laboratory “Neurosciences research laboratory”, Habib Bourguiba Hospital, Sfax.