Genetic Screening for Spinocerebellar Ataxia Genes in a Japanese Single-Hospital Cohort
Article information
Abstract
Objective
Diagnosis of sporadic cerebellar ataxia is a challenge for neurologists. A wide range of potential causes exist, including chronic alcohol use, multiple system atrophy of cerebellar type (MSA-C), and sporadic late cortical cerebellar atrophy. Recently, an autosomal-dominant spinocerebellar ataxia (SCA) mutation was identified in a cohort of patients with non-MSA-C sporadic cerebellar ataxia. The aim of this study is to genetically screen genes involved in SCA in a Japanese single-hospital cohort.
Methods
Over an 8-year period, 140 patients with cerebellar ataxia were observed. There were 109 patients with sporadic cerebellar ataxia (no family history for at least four generations, 73 patients with MSA-C, and 36 patients with non-MSA-C sporadic cerebellar ataxia) and 31 patients with familial cerebellar ataxia. We performed gene analysis comprising SCA1, 2, 3, 6, 7, 8, 12, 17, 31, and dentatorubro-pallidoluysian atrophy (DRPLA) in 28 of 31 non-MSA-C sporadic patients who requested the test. Familial patients served as a control.
Results
Gene abnormalities were found in 57% of non-MSA-C sporadic cerebellar ataxia cases. Among patients with sporadic cerebellar ataxia, abnormalities in SCA6 were the most common (36%), followed by abnormalities in SCA1 (7.1%), SCA2 (3.6%), SCA3 (3.6%), SCA8 (3.6%), and DRPLA (3.6%). In contrast, gene abnormalities were found in 75% of familial cerebellar ataxia cases, with abnormalities in SCA6 being the most common (29%). For sporadic versus familial cases for those with SCA6 abnormalities, the age of onset was older (69 years vs. 59 years, respectively), and CAG repeat length was shorter (23 vs. 25, respectively) in the former than in the latter (not statistically significant).
Conclusion
Autosomal-dominant mutations in SCA genes, particularly in SCA6, are not rare in sporadic cerebellar ataxia. The reason for the frequency of mutations in SCA6 remains unclear; however, the reason may reflect a higher age at onset and variable penetrance of SCA6 mutations.
Diagnosis of sporadic cerebellar ataxia is a challenge for neurologists. A wide range of potential causes exist, including chronic alcohol use, toxic agents, immune-mediated inflammation, vitamin deficiency, superficial siderosis, chronic central nervous system (CNS) infection, multiple system atrophy of cerebellar type (MSA-C), and sporadic cortical cerebellar ataxia (CCA) [1-4]. In Asian countries, including Japan, it is recognized that the most common sporadic cerebellar ataxia is MSA-C [4]. Recently, an autosomal dominant spinocerebellar ataxia (SCA) mutation has been recognized among non-MSA-C sporadic cerebellar ataxia [5-7]. Abele et al. [1] found that 18.8% of 80 patients with adult-onset sporadic cerebellar ataxia carry a disease-causing dominant SCA mutation, comprising seven SCA type 6, five Friedreich’s ataxia, two SCA3, and one SCA2. In Asian countries including China where Friedreich’s ataxia is rare, Wang et al. [8] found that 17.3% of 237 patients with sporadic cerebellar ataxia carry a disease-causing dominant SCA mutation, which includes 23 SCA3, nine SCA2, six SCA1, and three SCA6 mutations. Because SCA6 seems to have the latest disease onset among all types of SCA [9], mutations in the gene that causes SCA6 (CACNA1A) might be found most frequently in patients with sporadic cerebellar ataxia. To confirm this hypothesis, we genetically screened SCA genes in a Japanese single-hospital cohort.
MATERIALS & METHODS
This is a retrospective study. The initial inclusion criteria were any patients with cerebellar ataxia that were referred to our neurology clinic over an 8-year period. All patients were followed for 5 years in order to ascertain the clinical diagnosis. As the initial workup, all patients underwent standard neurological examination, cognitive screening tests comprising the Mini-Mental State Examination (0–30) [10] and the Frontal Assessment Battery (0–18) [11], brain imaging comprising magnetic resonance imaging (MRI) and single-photon emission computed tomography using [99mTc]-labeled L,L-ethyl cysteinate dimer. The exclusion criteria included degenerative neurologic diseases that potentially show cerebellar ataxia (progressive supranuclear palsy, prion disease, etc.), chronic alcohol use, Wernicke encephalopathy, hepatic encephalopathy, diphenylhydantoin intoxication, drug abuse, immune-mediated inflammation, vitamin deficiency, superficial siderosis, and chronic CNS infection [3] as determined through detailed history taking, blood tests, a nerve conduction study and examination of the cerebrospinal fluid, if necessary.
After the initial workup, 140 patients with cerebellar ataxia remained. The 140 patients were divided into 109 patients with sporadic cerebellar ataxia and 31 patients familial cerebellar ataxia as determined by a detailed history that covered over four generations (Figure 1). Among patients with sporadic cerebellar ataxia, only one showed consanguinity (his parents are cousins). Based on the published criteria [12], we excluded 73 patients who were diagnosed to have MSA-C. To ascertain the diagnosis, we performed an autonomic questionnaire and tests comprising a head-up tilt test, a urodynamics with sphincter electromyography [13], and a sleep study when sleep apnea/nocturnal stridor was suspected. The remaining 67 patients comprised 36 patients with nonMSA-C sporadic cerebellar ataxia and 31 patients with familial cerebellar ataxia. Among the 36 nonMSA sporadic cerebellar ataxia patients, 28 patients (80%) requested gene analysis. Among the 31 familial cerebellar ataxia patients, 24 patients (77%) requested gene analysis. Therefore, in the present study, a total 52 patients (28 non-MSA-C sporadic cerebellar ataxia and 24 familial cerebellar ataxia patients) served as study subjects, with those patients diagnosed with familial cerebellar ataxia serving as controls. Statistical analysis was conducted using Student’s t-test and Spearman’s rank correlation coefficient test.

Flow chart of patient recruitment. SCA: spinocerebellar ataxia, MSA-C: multiple system atrophy of cerebellar type.
RESULTS
Patient demographics
Using the clinical-imaging features of 52 cerebellar ataxia patients, 39 patients had cortical cerebellar degeneration (CCA, or pure cerebellar form) [14-16]. Of these patients, 23 had sporadic cerebellar ataxia, and 16 had familial cerebellar ataxia 16. None of the patients had neurological signs other than cerebellar ataxia (except for mild frontal executive dysfunction and mild positional vertigo, both reported in CCA [15]) by neurological examination and tests. None of the patients had atrophy/abnormal signals in brain areas other than the cerebellar hemisphere/vermis by MRI scans. Thirteen patients had non-CCA (sporadic 5, familial 8). Three of these 13 patients had extraocular movement abnormality, two had extrapyramidal sign, six had pyramidal sign, three had peripheral neuropathy, one had epilepsy, one had myoclonus, and one had decreased visual acuity. Brain MRI scans showed pontine atrophy in ten patients, and white matter lesions in one patient (Table 1). We performed gene analysis comprising ten genes [SCA1, 2, 3, 6, 7, 8, 12, 17, 31, and dentatorubro-pallidoluysian atrophy (DRPLA)] and CAG and TGGAA repeat length [17], the details of which are described in our previous study [18].

Gene analysis results and clinical-imaging features of spinocerebellar ataxia
Gene analysis results
In total, 34 (65%) of 52 cerebellar ataxia patients had gene abnormalities. SCA6 was the most common (17 patients, 32.7%), followed by SCA3 (6 patients, 11.5%), SCA31 (5 patients, 9.6%), SCA2 (2 patients, 3.8%), SCA1 (2 patients, 3.8%), SCA8 (1 patient, 1.9%), and DRPLA (1 patient, 1.9%). In the remaining patients, gene abnormalities were not identified. When divided by clinical-imaging subtypes, in non-CCA cases, gene abnormalities were found in 11/13 patients (85%). This is more than what was identified in CCA patients [23/39 patients (59%) (p < 0.05)]. Age at onset in CCA patients (mean, 55 years) was older than non-CCA patients (40 years), but the duration of the disease in CCA patients (mean, 12 years) was shorter than in non-CCA patients (15 years) (not statistically significant).
In non-MSA-C sporadic cerebellar ataxia, 16 (57%) of 28 patients had gene abnormalities. SCA6 was the most common (10 patients, 36%), followed by SCA1 (2 patients, 7.1%), SCA2 (1 patient, 3.6%), SCA3 (1 patient, 3.6%), SCA8 (1 patient, 3.6%), and DRPLA (1 patient, 3.6%). No abnormalities in SCA31 were observed. No gene abnormalities were identified in the remaining patients. When divided by clinical-imaging subtypes, in non-CCA patients, gene abnormalities were found in 5/5 (100%) patients. This is more than what was observed in CCA patients [11/23 patients (47.8%)]. SCA6 abnormalities were found in 10/23 patients (47.8%) with CCA (not statistically significant).
In familial cerebellar ataxia patients, 18 of 24 (75%) patients had gene abnormalities. SCA6 was most commonly mutated (7 patients, 29%), followed by SCA3 (5 patients, 20.8%), SCA31 (5 patients, 20.8%), SCA3 (1 patient, 3.6%), and SCA2 (1 patient, 4.2%). Gene abnormalities were not detected in remaining patients. All of the familial cerebellar ataxia cases might display autosomal dominant heredity. When divided by clinical-imaging subtypes, 5/7 (71%) nonCCA patients were shown to have gene abnormalities. This is almost the same as in CCA patients, which showed 13/17 (76%) of patients with abnormalities.
Among those with SCA6 (n = 17), if we compare sporadic (n = 10) versus familial cases (n = 7), age at onset was older (69 years vs. 59 years, respectively), and CAG repeat length was shorter (23 vs. 25, respectively) (not statistically significant) (Table 2).
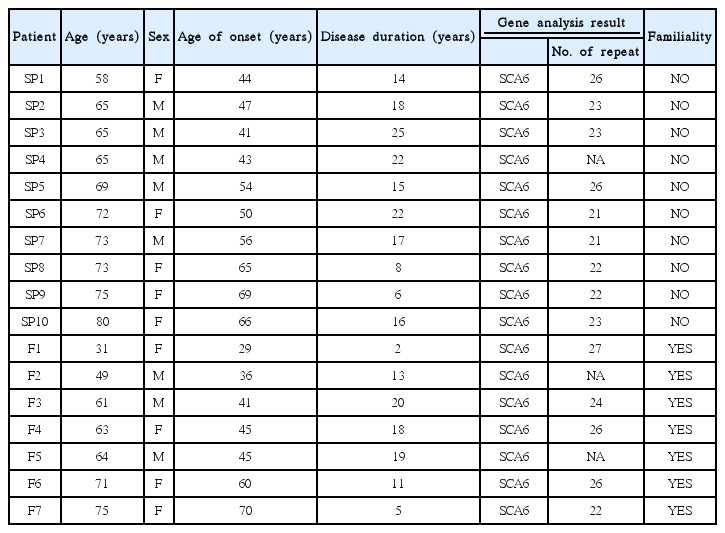
Comparison of sporadic and familial SCA6
DISCUSSION
The principal findings of this study can be summarized as follows: 1) Among non-MSA-C sporadic cerebellar ataxia, 57% of patients were found to have gene abnormalities. Among these, gene abnormalities in SCA6 were the most common (36%), followed by SCA1 (7.1%), SCA2 (3.6%), SCA3 (3.6%), SCA8 (3.6%), and DRPLA (3.6%). No abnormalities in SCA7, 12, or 31 were detected. Thus, autosomal-dominant SCA mutations, particularly in SCA6, are not rare in sporadic cerebellar ataxia. 2) Among those with SCA6 (n = 17), when comparing sporadic versus familial cases, age at onset was older (69 years vs. 59 years, respectively), duration of disease was longer (17 years vs. 13 years, respectively), and CAG repeat length was shorter (23 vs. 25, respectively); however, these differences were not statistically significant.
To the best of our knowledge, this is the first study to show the frequency of autosomal-dominant SCA mutations in sporadic cerebellar ataxia by testing ten genes. The hereditary nature of the disorder has not been well recognized; however, we show a high frequency rate (57%) of autosomal dominant SCA gene abnormalities in sporadic cerebellar ataxia. This is in contrast to previous reports, showing a frequency of 2–18.8% [1,2,5-9]. While we do not know the exact reason for this discrepancy, one explanation is the different method of gene testing that was used. For example, we tested ten genes, which could increase the rate of detecting gene abnormalities. Additionally, familial relations may have been underreported in our subjects. Another explanation for the high frequency of SCA6 abnormalities is that this cohort had a higher age at onset and a variable penetrance of SCA6 abnormalities. This may also be attributable to the ethnicity or the geographical clustering of the families. SCA6 is the most common SCA subtype in Japan.
Because SCA6 seems to have the latest disease onset among all types of cerebellar ataxia [9], mutations in the gene that causes SCA6 (CACNA1A) might be found more frequently in sporadic cerebellar ataxia. The results of the present study confirmed this assumption, showing that SCA6 was the most common (36%) dominant SCA mutation in sporadic cerebellar ataxia. This finding is also noted by Abele et al. [1].
Among those with SCA6, comparing sporadic versus familial cases, the age at onset was older (69 years vs. 59 years), and CAG repeat length was shorter (23 vs. 25, respectively) in the former. Thus, in SCA6, sporadic cases might have milder clinical-genetic feature than those of familial cases. However, this requires further clarification with a larger number of patients. This seems in accordance with the notion that developing SCA6 without a hereditary component could be due to a new mutation, or because of false paternity, incomplete penetrance and/or death of the transmitting parent before onset of clinical symptoms [1-3]. The present study results also prompted the assumption that there is an overlap between SCA6 and sporadic CCA [14-16,19]. This is because SCA6 usually has the imaging findings and pathologic substrate confined to the cerebellar cortex.
Limitations of this study include that some of our patients underwent gene analysis at other institutions, and therefore, the number of triplet repeats is missing. This has the potential of interfering with the results of the clinical-genetic relationship. Additionally, we were unable to analyze additional SCA genes (for example, SCA11). Selection bias, such as the ambiguity of inclusion or exclusion and incomplete family history might also contribute to the discrepancy between the frequencies of gene abnormality in the present study and those in the previous studies. Although the number of our study subjects was small, our study results clearly indicate that autosomal-dominant SCA mutation, particularly SCA6, is not rare in SCA. This finding sheds light on proper patient care and the ability of early intervention of this disorder in the future.
In conclusion, autosomal-dominant SCA mutations, particularly SCA6, is not rare in sporadic cerebellar ataxia. The exact reason remains unclear, presumably reflecting higher age at onset and variable penetrance of SCA6.
Notes
Conflicts of Interest
The authors have no financial conflicts of interest.