Prospective Characterization of Cognitive Function in Typical and ‘Brainstem Predominant’Progressive Supranuclear Palsy Phenotypes
Article information

Abstract
Objective
Clinicopathological studies over the last decade have broadened the clinical spectrum of progressive supranuclear palsy (PSP) to include several distinct clinical syndromes. We examined the cognitive profiles of patients with PSP-Richardson’s syndrome (PSP-RS) and two atypical ‘brainstem predominant’ PSP phenotypes (PSP-parkinsonism, PSP-P; and PSP-pure akinesia with gait freezing, PSP-PAGF) using a comprehensive neuropsychological battery.
Methods
Fourteen patients diagnosed as PSP-RS, three patients with PSP-P and four patients with PSP-PAGF were assessed using a comprehensive battery of neuropsychological tests.
Results
The typical PSP-RS subgroup demonstrated greater impairments in processing speed [t(19) = -4.10, p = 0.001 (d =1.66)] and executive function [t(19) = -2.63, p = 0.02 (d = 1.20)] compared to the ‘brainstem predominant’ PSP phenotype.
Conclusion
This is the first prospective study to demonstrate that PSP-RS and ‘brainstem predominant’ PSP phenotypes can be differentiated on cognitive grounds. These differences correspond with variations in pathological profiles reported in the literature.
Clinicopathological studies over the last decade have broadened the clinical spectrum of progressive supranuclear palsy (PSP). A new nosology has emerged that includes the ‘classic’ or typical PSP-Richardson’s syndrome (PSP-RS) and a variety of ‘atypical’ PSP clinical syndromes [1,2]. The atypical PSP phenotypes are differentiated from PSP-RS by distinct clinical features as well as variations in the distribution of PSP-tau pathology [2-4]. To date, the literature has largely focused on identifying pathological and neurological variations among the PSP phenotypes. The aim of this study was to determine whether the cognitive profile of typical PSP (PSP-RS) can be differentiated from the most pathologically distinct ‘brainstem predominant’ PSP phenotypes [3], namely, PSP-parkinsonism (PSP-P) and PSP-pure akinesia with gait freezing (PSP-PAGF). Despite having less severe brainstem pathology than PSP-RS, PSP-P, and PSP-PAGF are referred to as ‘brainstem predominant’ PSP phenotypes because the pathology is more concentrated in the brainstem relative to some other atypical phenotypes that have more severe cortical pathology [4-10].
Neuropsychological studies have consistently demonstrated the presence of cognitive symptoms in the majority of cases of typical PSP [11-15]. PSP-RS is associated with a specific cognitive betweenprofile, which includes cognitive slowing, deficits in attention, and early and severe frontal executive dysfunction with difficulties in allocating attentional resources, problems with planning, shifting concepts and prominent retrieval-based memory deficits [11-15]. These cognitive deficits have been linked to subcortical pathology and associated frontal deafferentation [16] as well as damage to cortical frontal regions and underlying white matter tracts [11,17].
To date, only two studies have prospectively examined the neuropsychological profiles of atypical PSP phenotypes [7,18]. Both of these studies compared PSP-P with typical PSP-RS. Although they found no significant differences between the phenotypes in general cognition or executive function, neither study comprehensively examined cognition. This has left unanswered, the question of whether clinicopathological distinctions between these PSP groups extend to differences in cognitive profiles in a prospective cohort. Clarifying this issue may aid earlier and more accurate differentiation of classic and ‘brainstem predominant’ PSP phenotypes.
To this end, this study utilized a comprehensive neuropsychological examination to prospectively investigate the severity of cognitive changes in typical PSP and the ‘brainstem predominant’ PSP phenotypes. Given the greater pathological involvement of the basal ganglia, diencephalon, brainstem, cerebellum and frontal cortical structures in PSP-RS compared to the ‘brainstem predominant’ PSP phenotypes (PSP-P, PSP-PAGF), it was predicted that the PSP-RS group would exhibit more severe deficits in processing speed, attention and executive function than the ‘brainstem predominant’ PSP phenotype group.
MATERIALS & METHODS
Participants
Fourteen patients with PSP-RS, 3 patients with PSP-P, and 4 with PSP-PAGF were recruited from a specialist movement disorder clinic at a tertiary referral hospital in Melbourne, Australia. Participants were diagnosed by an experienced movement disorder specialist (D.W.) according to the diagnostic criteria for PSP phenotypes [19]. The inclusion criteria for PSP-RS were early falls, cognitive dysfunction, eye movement abnormalities, and postural instability; PSP-P included asymmetric parkinsonian symptoms (rigidity, bradykinesia and in some cases tremor), non-axial dystonia and modest response to levodopa; and PSP-PAGF included an early history of freezing of gait or hypophonia of gradual onset without limb rigidity and tremor and no sustained response to levodopa, dementia or eye movement abnormalities. Patients with history of major focal neurological or psychiatric disorders (history of stroke, severe depression, or psychosis), major head injury, drug or alcohol abuse, and global cognitive decline (as indicated by the Mini Mental State Examination score < 24) were excluded. No participants had their diagnosis changed during the two-year follow-up period, and in all participants who have died since study commencement (n = 5), post-mortem findings have confirmed the study neurologist’s clinical diagnosis (PSP-RS).
Materials and procedures
Clinical examination
All participants underwent systematic neurological examination that included the Progressive Supranuclear Palsy Rating Scale [20] and the Hoehn and Yahr Stage of Illness Rating Scale (HYRS) [21].
Neuropsychological battery
All participants completed a comprehensive neuropsychological examination that assessed six cognitive domains: processing speed, memory, language, working memory, visuospatial function, and executive function (Table 1 for list of tests).

Neuropsychological tests and corresponding baseline measures
To minimize the influence of motor, speech and oculomotor dysfunction on test performance, baseline control comparisons, modified scoring and presentation procedures were used (Table 1).
All patients gave written informed consent to the study, which was approved by The Alfred Hospital Human Research Ethics Committee (HREC reference: 66/09).
Statistical analyses
Nonparametric Mann-Whitney tests were used to compare the clinical and demographic characteristics of the groups. Cognitive domain indices were derived by averaging the z-scores of the tests within each cognitive domain by group [22]. For 5 of the 6 indices, the aforementioned assessment modifications were incorporated into the index, allowing between-group analysis with t-tests for unpaired samples. The language index required covariation of baseline speed, so analysis of covariance was conducted for this index. Due to the limited sample size, no corrections for multiple comparisons were made.
Standardized scores were calculated for each cognitive test using published normative data. Impairment was classified relative to the normative mean as mild [≥ 1.5 standard deviations (SD)], moderate (≥ 2 SD), or severe (≥ 3 SD) [23].
RESULTS
Demographic and clinical data
As shown in Table 2, there were no significant differences between the typical and ‘brainstem predominant’ PSP phenotypes on age [mean (SD) = 69.93 (5.66) vs. 67.67 (15.63); p = 0.40], education [mean (SD) = 10.79 (2.69) vs. 13.33 (4.16); p = 0.26], levodopa medication (on levodopa = 6/14 vs. 4/7; p = 0.10; Fisher’s test) or level of clinical disability (PSPRS: p = 0.09; HYRS: p = 0.16). Thus, the groups were well matched with respect to the demographic variables and disease severity.
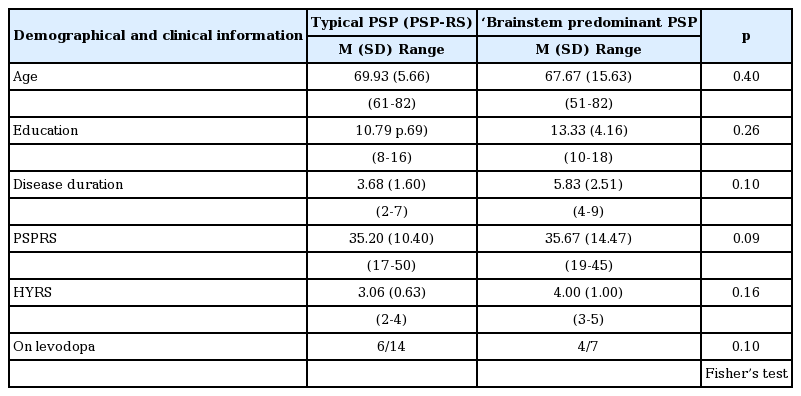
Demographic and clinical characteristics of the PSP phenotypes
Not surprisingly, the PSP-RS group showed a trend towards shorter disease duration relative to the ‘brainstem predominant’ PSP phenotypes (3.7 vs. 5.8; p = 0.10).
Neuropsychological performance
As shown in Table 3, there were significant differences between the groups on processing speed, [t(19) = -4.10, p = 0.001, d = 1.66], with the PSPRS group showing significantly slower general processing speed (mean = -0.36, SD = 0.89) than the ‘brainstem predominant’ PSP group (mean = 0.76, SD = 0.35). Similarly, the PSP-RS group performed significantly worse (mean = -0.27, SD = 0.65) than those in the ‘brainstem predominant’ PSP group (mean = 0.54, SD = 0.69) on the executive function index [t(19) = -2.63, p = 0.02, d = 1.20]; the effect size of these differences was large. In contrast, no significant group differences were observed for the memory, [t(19) = -1.01, p = 0.32, d = 0.47], language [t(19) = 3.83, p = 0.42, d = 0.92], working memory [t(19) = 0.41, p = 0.19, d = 0.62], and visuospatial function indices, [t(19) = -1.13, p = 0.28, d = 0.30].
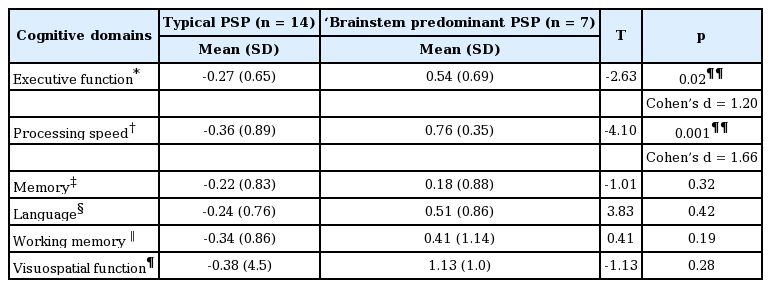
Between group comparisons [means (SD)] of the typical and ‘brainstem predominant’ PSP phenotypes on cognitive indices and the mean performance of the ‘brainstem predominant’ PSP phenotypes relative to age- and educationstratified normative data
As shown in Table 4, supplementary analyses comparing PSP-P and PSP-PAGF from a clinical perspective revealed mild to moderate impairments on measures of processing speed, verbal memory and fluency for all individuals in the PSP-P group. In contrast, no individuals with PSP-PAGF demonstrated clinically significant impairment (≤ 1.5 SD) in any cognitive domains.

Mean performances of the PSP phenotypes relative to age- and education-stratified normative data
DISCUSSION
This study shows that typical PSP-RS has a cognitive profile that is distinct from the cognitive profile of the ‘brainstem predominant’ PSP phenotypes (PSP-P and PSP-PAGF). Specifically, we found significantly greater executive function deficits and significantly slower speed of information processing in the PSP-RS subgroup compared to the ‘brainstem predominant’ PSP phenotypes. It is unlikely that these findings were primarily due to inter-group variations in speech or motor deficits, as the groups were well matched with respect to severity, and the assessment included baseline control comparisons and measurement modifications to mitigate the influence of speech or motor deficits.
The evidence of more pronounced psychomotor slowing and executive dysfunction in the PSP-RS subgroup is largely in accordance with reported pathological differences between the typical and ‘brainstem predominant’ PSP phenotypes [3,4,8]. Specifically, the greater executive dysfunction documented in the current PSP-RS cohort corresponds well to reports of greater frontal pathology in this group [5-8]. The more pronounced psychomotor slowing also corresponds well to the more severe subcortical pathology in the PSP-RS group [4].
Clinically-driven consideration of cognition in the ‘brainstem predominant’ PSP phenotypes indicated that it may be possible to identify discrete cognitive profiles in PSP-P and PSP-PAGF. Specifically, none of the individuals in the PSP-PAGF group demonstrated clear evidence of cognitive impairment on any domain of function, whereas all of the individuals in the PSP-P group showed mild to moderate impairment in processing speed, memory and fluency.
PSP-RS has a shorter naturally occurring disease course than the ‘brainstem predominant’ PSP phenotypes, but the groups in this study were well matched in terms of disease severity. It is therefore unsurprising that the PSP-RS group trended (p = 0.10) toward having a shorter mean disease duration (3.68 years) than the ‘brainstem predominant’ PSP phenotypes (5.33; 5.50 years). This trend is unlikely to be the cause of the group differences in cognitive ability, as the PSP-RS group performed more poorly on cognitive assessment while being earlier in their disease duration than the ‘brainstem predominant’ PSP phenotypes. Given that the groups were well matched in terms of disease severity, it seems probable that the relatively greater impairment in the PSP-RS group represents a real group difference between these phenotypes.
The relatively small size of the patient sample in the current study and the fact that clinical diagnoses were made in vivo limits the interpretation of the results. However, all of the cases included in the current study have been seen at a tertiary center with a high rate of clinical diagnostic accuracy confirmed by pathological examination. Moreover, current significant findings together with the large effect sizes despite the small size of the PSP groups suggest that the reported group differences in cognition are clinically meaningful. The small sample size may, however, have resulted in insufficient statistical power to identify subtler group differences in other cognitive domains. Future work with larger cohorts would be able to address this issue.
The field of PSP research is moving away from considering PSP as a single clinical entity [1,3,6,7]. This prospective study offers the first clear evidence that typical PSP-RS can be differentiated from two ‘brainstem predominant’ PSP phenotypes (PSP-P and PSPPAGF) on the basis of cognitive ability. Specifically, individuals with PSP-RS have more impaired executive functioning and processing speed than individuals with the ‘brainstem predominant’ PSP phenotypes. The current findings provide preliminary evidence that PSP-RS and ‘brainstem predominant’ PSP phenotypes may be differentiated by the severity of cognitive deficits. Further studies, with larger sample sizes are clearly needed, but this study suggests that such work may be clinically fruitful.
Notes
Conflicts of Interest
The authors have no financial conflicts of interest.
Acknowledgements
We would like to thank Dr. Will Lee for assisting in the collection of the clinical data used in this study. We would like to thank Prof. Catrina McLean for providing the pathological diagnoses, and we are grateful for the assistance of Fairlie Hinton at the Victorian Brain Bank Network.