Fragile X-Associated Tremor/Ataxia Syndrome: An Illustrative Case
Article information

Fragile X-associated tremor ataxia syndrome (FXTAS) is an uncommon neurodegenerative disorder caused by a premutation of the CGG repeat expansion within the 5' UTR of the fragile-X mental retardation (FMR1) gene. Neurological manifestations of FXTAS include action tremor, ataxia, parkinsonism, cognitive impairment, peripheral neuropathy, and autonomic dysfunction and typically occur in middle-aged men [1]. Here, we report a patient with FXTAS who presented typical clinical symptoms and structural and functional imaging features as an illustrative case report.
A 60-year-old man who presented with action hand tremors and cognitive impairment since the age of 53 visited our hospital. Bilateral hand postural tremors, which were aggravated during writing or spoon feeding, was the first noted symptom. There was no resting tremor initially, and the action tremor was not severe enough to interrupt his daily life. His family members also reported that he had decreased desire, loss of conversation ability, and slight memory disturbance with the onset of tremor. At the age of 54, resting tremors presented. The patient had taken propranolol and primidone for several years under the diagnosis of essential tremor, but there was no clinical effect. The tremor gradually progressed until he was unable to feed himself or write due to severe action tremors by the age of 60. His memory disturbance also had progressed and became more apparent when he could not remember simple appointments with his family members.
Upon admission, no abnormal findings were observed in the cranial nerve or in motor and sensory function tests. On parkinsonism examination, mild bradykinesia was observed in all limbs. The resting tremor was mild in degree, and the postural and action tremors were moderate in degree and bilaterally symmetrical in the arms. The finger-to-nose test and heel-to-shin test showed mild dysmetria. Deep tendon reflexes were hypoactive in all limbs, and there were no pathologic reflexes. His had a wide-based gait, but his arm swing and walking speed were preserved. Tandem gait was impaired. There was no postural instability. His Mini-Mental State Examination score, Montreal Cognitive Assessment score, and Global Deterioration Scale score were 27, 25, and 3, respectively. There was no significant depression noted. During the Seoul Neuropsychologic Screening Battery, his performance was below normal in Word Stroop Tests, Digit Symbol Coding, Trail Making Tests, suggesting frontal/executive dysfunction. He denied a family history of parkinsonism, dementia, tremor, or other neurological diseases.
Laboratory tests indicated no abnormal findings with respect to complete blood count, electrolyte levels, and kidney and liver functions. Brain MRI showed high signal intensities in the middle cerebellar peduncle (MCP), periventricular/deep white matter on fluid attenuated inversion recovery (FLAIR) image and diffuse brain atrophy (Figure 1A-D). There were no definite abnormalities in [18F] N-3-floropropyl-2β-carboxymethoxy-3β-(4-iodophenyl)-nortropane ([18F]-FP-CIT) positron emission tomography (PET) or in [18F] fluorodeoxyglucose ([18F]-FDG) PET imaging (Figure 1E and F).
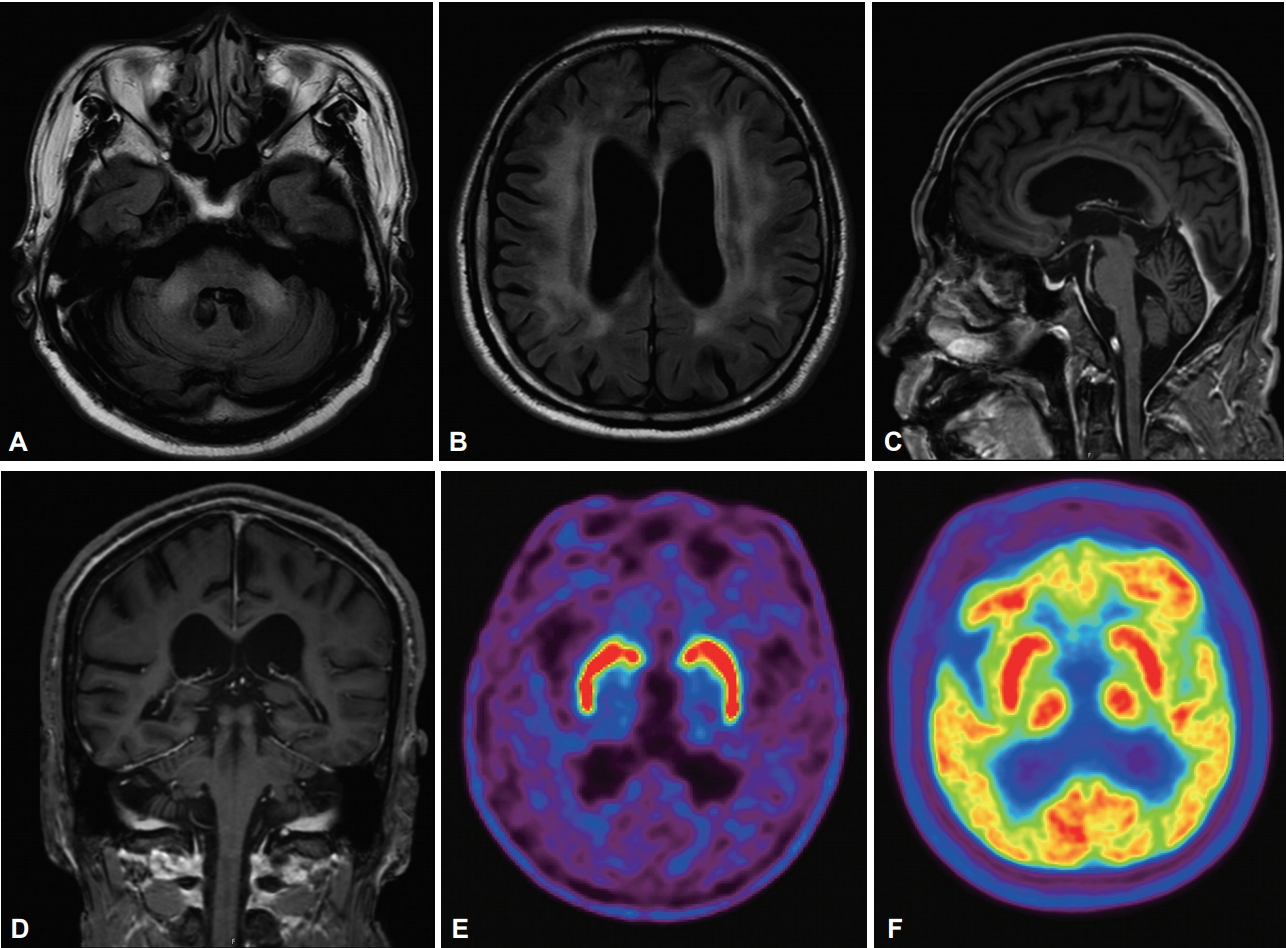
The axial section of the fluid attenuated inversion recovery brain MRI shows high signal intensity in the bilateral middle cerebellar peduncle and periventricular/deep white matter (A and B). Sagittal and coronal sections show prominent subarachnoid space and atrophy of the bilateral cerebellar hemisphere (C and D). Preserved uptakes in [18F]-FP-CIT PET (E) and preserved glucose metabolism in [18F]-FDGPET (F).
We performed genetic testing for the spinocerebellar ataxia (SCA) and FMR1 genes. The SCA1, SCA2, SCA3, SCA6, SCA7, SCA8, and SCA17 genes showed no mutation. However, the FMR1 gene showed premutation with a CGG repeat number of 100 (normal < 55). We diagnosed this patient as having FXTAS.
We report a typical FXTAS case with FMR1 gene premutation with a repeat number of 100. FXTAS, a neurodegenerative disorder, is associated with carriers of the FMR1 gene permutation (CGG repeat number range: 55–200). Fragile X syndrome is caused by FMR1 full mutation (> 200 CGG repeat numbers), which leads to hypermethylation and transcriptional silencing of the fragile X mental retardation protein (FMRP). The absence of FMR1 mRNA and FMRP in fragile X syndrome results in cognitive impairment and autistic features in male children [1]. However, premutation of the FMR1 gene is associated with elevated levels of FMR1 mRNA. On autopsy, FMR1 mRNA can be observed in inclusion bodies [2,3]. These findings suggest that an increased density of FMR1 mRNA is associated with FXTAS neurotoxicity to reveal various symptoms. Due to the gain-of-function mechanism of FXTAS, neurodegeneration typically occurs in middleaged men [1].
The incidence of premutation in the FMR1 gene is 1 in 800 men and 1 in 250 women, but the symptoms are present in 20% of women and 30–40% of men carriers. The penetrance rate increases with age, and the average age of onset in male carriers is in the early sixties [4,5]. Symptom onset and severity are related to the number of CGG repeat numbers; a higher repeat number is associated with earlier symptom onset, more severe symptoms and a short life expectancy [6].
Characteristic brain MRI findings in FXTAS patients are atrophy and T2/FLAIR high signal intensities in both MCPs. This is also an important clue for diagnosing FXTAS [7]. Diagnostic criteria for FXTAS, suggested by Jacquemont et al. [4] in 2003, include clinical symptoms such as gait ataxia and intention tremor with characteristic MRI findings, including T2/FLAIR high signal intensity in the MCP, which are of a high diagnostic value. However, clinical symptoms should always be considered simultaneously when diagnosing FXTAS because high signal intensities of the MCPs are characteristic of but not specific to FXTAS. Other neurodegenerative diseases affecting pontocerebellar fibers, such as multiple system atrophy (MSA) or SCA, have also been observed. The classic symptoms of FXTAS are action tremor and gait ataxia noted after the age of 50 years. There are more diverse clinical symptoms, such as peripheral neuropathy, parkinsonism and hypermotor symptoms. Because of this symptom diversity, FXTAS is often misdiagnosed as an essential tremor, Parkinson’s disease, or MSA. In our case, there was both hand action tremor with gait and limb ataxia, and MRI showed MCP atrophy and T2 high signal intensity; therefore, we presumed our patient to have FXTAS and confirmed the diagnosis with a genetic test for the FMR1 gene.
FXTAS is a newly recognized neurodegenerative disease that has diverse neurological symptoms. It can be misdiagnosed as other neurodegenerative diseases. The present case shows characteristic tremor, ataxia, and MRI findings for FXTAS. This illustrative case suggests that FXTAS should be considered as a differential diagnosis in middle-aged men who show action tremor, parkinsonism or cognitive dysfunction with characteristic MRI findings of bilateral T2 high signal intensity in the middle cerebellar peduncles, even if there is no family history of the typical phenotype.
Notes
Conflicts of Interest
The authors have no financial conflicts of interest.
Acknowledgements
This work was supported by a grant from the Korea Healthcare Technology R&D Project, Ministry of Health and Welfare, Republic of Korea (HI17C0328).