Extensive Leukoencephalopathy in Spastic Paraplegia Type 4: Possible Role of Cerebral Autosomal Arteriopathy With Subcortical Infarcts and Leukoencephelopathy
Article information

Abstract
Despite recent advances in next-generation sequencing, the underlying etiology of adult-onset leukoencephalopathy has been difficult to elucidate. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is a representative hereditary adult-onset leukoencephalopathy associated with vasculopathy. Leukoencephalopathy in spastic paraplegia type 4 (SPG4) is known to be rare, but it might be underestimated because most spastic paraplegia with leukoencephalopathy is rarely considered. We report a case of co-occurring SPG4 and CADASIL. A 61-year-old male presented with sudden visual impairment after a headache. He showed a spastic gait and had a family history with similar symptoms. An SPG4 gene mutation and a pathogenic variant in the NOTCH3 gene were found. This case shows that the diverse and complex clinical manifestations of patients with extensive leukoencephalopathy are related to more than one gene mutation. We also suggest the necessity for relevant genetic tests in the diagnosis of adult-onset leukoencephalopathy.
Hereditary spastic paraplegias (HSPs) are a group of clinically and genetically heterogeneous disorders with progressive spastic gait disturbance and weakness in the lower limbs [1]. Spastic paraplegia type 4 (SPG4) caused by the SPAST gene mutation is the most common subtype of HSP. Extensive white matter (WM) changes in SPG4 are not frequent [1,2].
We report a patient with extensive WM lesions and progressive spastic paraplegia of SPG4 having concurrent mutations in NOTCH3 and SPAST.
CASE REPORT
A 61-year-old male presented with visual disturbance after a severe pulsating headache. He had developed progressive spastic gait five years ago, and his mother and maternal cousins had also shown gait disturbances (Supplementary Figure 1 in the online-only Data Supplement). The differential diagnosis of progressive myelopathy with paraplegic gait was made for other causes, including genetic testing. DNA analysis of the patient revealed a five-base deletion in exon 9 of the SPG4 gene (c.1215_1219del, p.N405fs; SPAST gene) (Figure 1A), which was a previously reported mutation. As an SPG4-related imaging feature, a thin corpus callosum was suspicious, but magnetic resonance imaging (MRI) showed no obvious atrophy in the cerebellum or spinal cord.
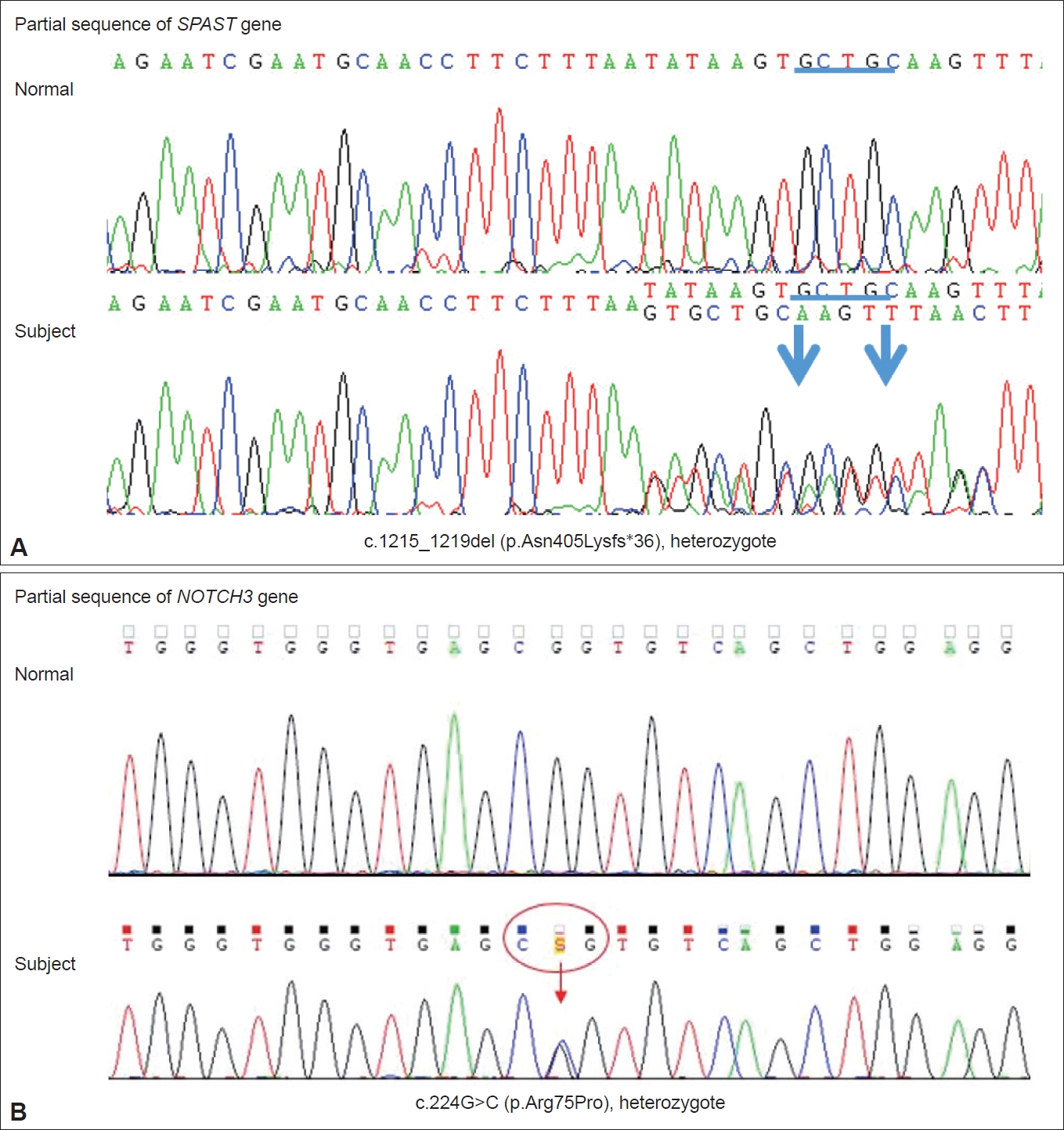
Sanger sequencing chromatograms. The heterozygous pathogenic mutations in the SPAST gene [c.1215_1219del (p.N405fs)] (A) and NOTCH3 gene [c.224G>C (p.Arg75Pro)] (B) were confirmed. Chromatograms from a normal control subject are also shown for comparison. The blue underline and arrows in Figure 1A represent the deletion location and both ends of the deletion breakpoint, respectively. The red circle and arrow in Figure 1B indicate the codon affected by the missense mutation and the corresponding variant location, respectively.
On admission for visual disturbance and severe headache, neurologic examination revealed left homonymous hemianopsia. Brain MRI showed focal acute infarctions in the right parietal cortices, but there was no definite abnormality corresponding to left homonymous hemianopsia, and the visual disturbance gradually improved over two days after admission to the hospital. Electroencephalogram did not show epileptiform discharge. Detailed etiologic work-up for ischemic stroke, including magnetic resonance angiography, echocardiography, and 24-hour Holter monitoring, did not reveal a specific etiology. He had a history of recurrent headache compatible with migraine. Given the accompanying migrainous headache and history of migraine, we presumed that the causes of the transient visual field defect and acute cerebral infarction would be atypical migraine aura and migrainous infarction, respectively. Interestingly, brain MRI further showed fronto-parieto-occipital confluent hyperintense lesions in the cerebral WM, indicating leukodystrophy-like lesions (Figure 2). On detailed history-taking, he had been experiencing word retrieval failures and difficulty remembering conversations or the names of people for several months. However, the Mini-Mental State Examination and Montreal Cognitive Assessment scores were 28 and 22, respectively, and there were no significant difficulties in activities of daily living ascribed to cognitive impairment. Because of the abnormal MRI findings and clinical features such as migraine, we additionally performed sequencing of the NOTCH3 gene despite unclear family histories related to CADASIL. He had a heterozygote of a pathogenic variant, c.224G>C (p.Arg75Pro) in exon 3 of the NOTCH3 gene (Figure 1B). SPG4- and/or CADASIL-relevant symptoms and signs of the patient are summarized as a Venn diagram in Supplementary Figure 2 (in the online-only Data Supplement).
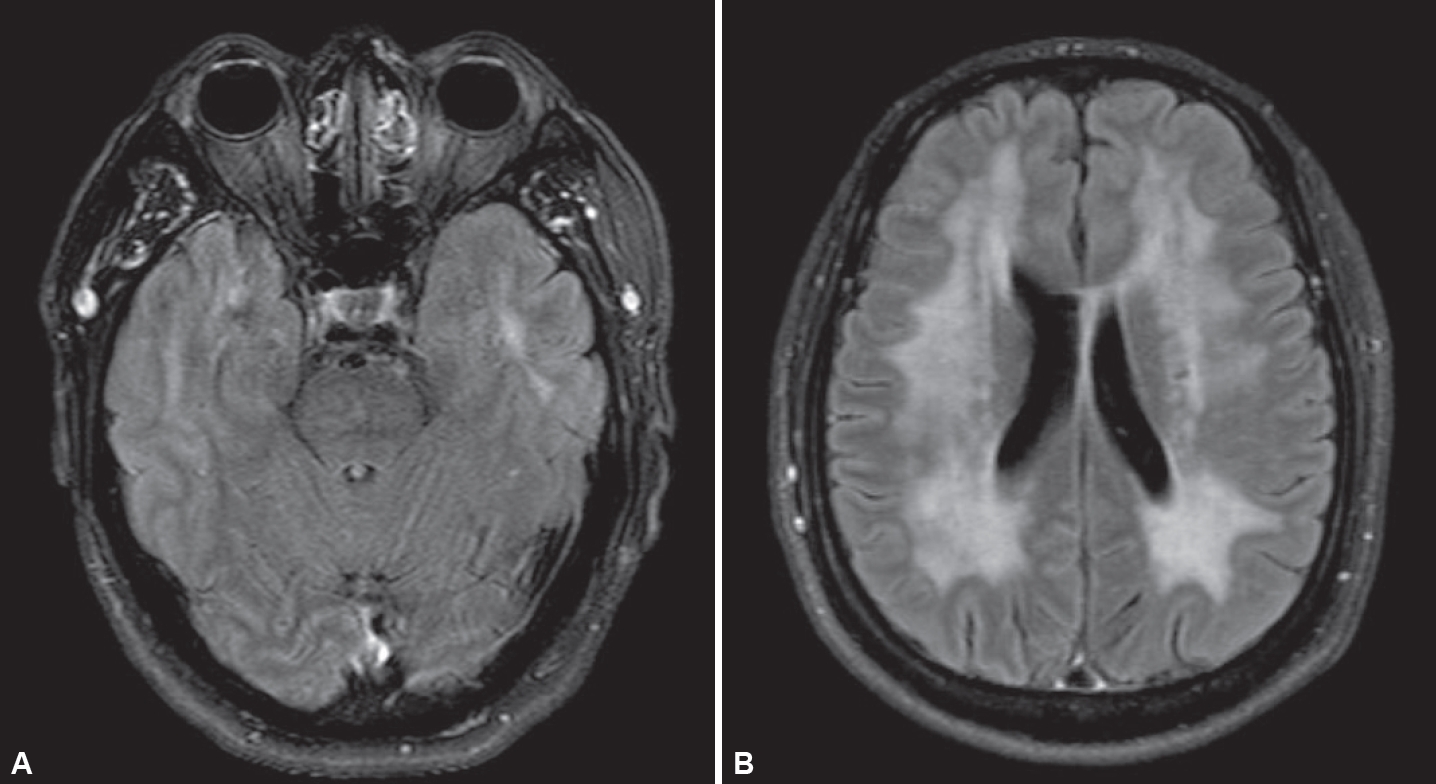
Brain magnetic resonance imaging (MRI). A: Axial fluid-attenuated inversion recovery (FLAIR) image showing high signal intensity in the bilateral anterior temporal lobes. B: Axial FLAIR image showing confluent high signal intensity involving bilateral cerebral white matter.
His 31-year-old daughter did not show any symptoms, but his 29-year-old son often complained of a mild episodic headache. Genetic tests were also performed with his daughter and his son. The daughter did not show any pathogenic or likely pathogenic variant, but his son had the same pathogenic variants in the NOTCH3 and SPAST genes as his father.
DISCUSSION
This case was unusual and unique in the identification of two genetic mutations associated with the various clinical phenotypes of the patient. The patient initially showed gait disturbance, which was related to SPG4 resulting from heterozygous mutations in the SPAST gene (2p22.3).
HSPs are a group of clinically and genetically heterogeneous disorders with progressive spastic gait disturbance and weakness in the lower limbs [1]. SPG4 accounts for 40% of autosomal dominant HSP cases. Although most cases of SPG4 are classified as pure HSP with isolated pyramidal signs (spasticity, hyperreflexia, etc.), complicated types of HSP have been reported. Manifestations of complicated HSP have included cognitive impairment, epilepsy, widespread effects in gray and WM, developmental anomalies, ataxia, and seizures [1,2]. Nonspecific WM lesions in the brain are frequent in complicated phenotypes and are occasionally also described in pure HSP forms (e.g., SPG4 and SPG5) [2].
Initially, the patient presented with gait disturbance with spasticity and was diagnosed with HSP through SPAST gene sequencing. His cognitive change occurred slowly afterward, and WM lesions were considered a phenotype of complicated HSP. However, migrainous headache and having the temporal pole involved in the WM lesions suggested the differential diagnosis of diseases other than HSP. NOTCH3 genetic testing was performed because the brain MRI results were compatible with CADASIL [3].
In several studies, the rate of multiple molecular genetic diagnoses has been reported in 3.2% to 7.2% of cases in which a molecular diagnosis is made, and it does not seem to be very rare [4-9]. Furthermore, a large-scale retrospective study of 7,374 referrals suggested a possible underrecognition of pathogenic variants at multiple molecular loci even during an interpretation of whole exome sequencing [9]. The suggestion was based on the finding that the observed proportion of patients with a diagnosis who had multiple molecular diagnoses was significantly lower than that expected by chance alone [9]. Therefore, even if a genetic diagnosis is confirmed in a patient, an additional molecular genetic diagnosis needs to be considered if the patient has a clinical feature that is more compatible with another genetic disorder.
In conclusion, we report a rare case in which two genetic variants with a SPAST mutation (c.1215_1219del, p.N405fs) and NOTCH3 gene mutation (c.224G>C, p.Arg75Pro) were found simultaneously. Additionally, extensive WM changes in HSP might require consideration of other causes of adult-onset leukoencephalopathy, including CADASIL, rather than the HSP itself [10].
Supplementary Materials
The online-only Data Supplement is available with this article at https://doi.org/10.14802/jmd.21091.
Supplementary Figure 1.
Pedigree chart. This pedigree shows the phenotype between the patient and family members. *A case of sequencing of the SPAST and NOTCH3 genes; †A case of heterozygous pathogenic missense mutation in exon 3 of the NOTCH3 gene [c.224G>C (p.Arg75Pro)] and heterozygous pathogenic five-base deletion in exon 9 of the SPAST gene [c.1215_1219del, p.N405fs].
Supplementary Figure 2.
A Venn diagram summarizing SPG4- and/or CADASIL-relevant symptoms and signs in the patient. WMHs, white matter hyperintensities; SPG4, spastic paraplegia type 4; CADASIL, Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy.
Notes
Ethics Statement
This work was approved by the ethics committees of the Inje University Busan Paik Hospital (IRB No. 2021-04-015) and written informed consent was obtained from the patient.
Conflicts of Interest
The authors have no financial conflicts of interest.
Funding Statement
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2020R1G1A1008446).
Author Contributions
Conceptualization: Jin Ho Jung, Eun Joo Chung, Seong-il Oh. Data curation: all authors. Formal analysis: all authors. Funding acquisition: Seong-il Oh. Investigation: Jin Ho Jung, Eun Joo Chung, Seong-il Oh. Methodology: Jin Ho Jung, Eun Joo Chung, Seong-il Oh. Project administration: Eun Joo Chung, Seong-il Oh. Resources: Eun Joo Chung, Seong-il Oh. Supervision: Eun Joo Chung, Seong-il Oh. Writing—original draft: Jin Ho Jung, Eun Joo Chung, Seong-il Oh. Writing—review & editing: all authors.
Acknowledgements
We thank all the participants of this research.