Effect of Chelation Therapy on a Korean Patient With Brain Manganese Deposition Resulting From a Compound Heterozygous Mutation in the SLC39A14 Gene
Article information

Abstract
Mutations in the manganese transporter gene SLC39A14 lead to inherited disorders of manganese metabolism. Chelation therapy with edetate calcium disodium (SLC39A14 deficiencyTA) is known to effectively reduce manganese deposition. We describe the first identified Korean case of SLC39A14-associated manganism and the treatment response to a 5-year chelation therapy. An 18-year-old female presented with childhood-onset dystonia. Magnetic resonance imaging showed T1 hyperintensity throughout the basal ganglia, brainstem, cerebellum, cerebral and cerebellar white matter, and pituitary gland. Blood manganese levels were elevated, and whole-exome sequencing revealed compound heterozygous mutations in SLC39A14. Treatment with intravenous CaNa2EDTA led to a significant reduction in serum manganese levels and T1 hyperintensities. However, her dystonia improved insignificantly. Hence, early diagnosis of this genetic disorder is essential because it is potentially treatable. Even though our treatment did not significantly reverse the establish deficits, chelation therapy could have been more effective if it was started at an earlier stage of the disease.
Inherited disorders of manganese (Mn) metabolism have recently been discovered [1]. Mutations in the SLC30A10 and SLC39A14 genes, which encode Mn transporters, lead to hypermanganesemia and Mn accumulation in the brain [1,2]. Chelation with edetate calcium disodium (CaNa2EDTA) has been effectively used to reduce Mn accumulation [3]. Here, we describe the first Korean case of brain Mn accumulation due to SLC39A14 mutations and the treatment response to chelation therapy for 5 years.
CASE REPORT
An 18-year-old female had developed twisting postures in her feet with tip-toe gait at the age of five. Her dystonia progressively worsened and spread to the neck, trunk, and limbs. Two years after its onset, she was unable to walk and became wheelchair-bound. Her perinatal history was unremarkable, and her early development was normal. There was no history of dystonia in her family and no environmental exposure to heavy metals. She did have a history of iron-deficiency anemia and menorrhagia. Her dystonia often worsened during the period prior to menstruation.
Neurological examination revealed dysarthria and generalized dystonia involving the face, neck, trunk, and limbs. She displayed an open-mouthed dystonic smile and slow writhing movements of her limbs. Her deep tendon reflexes were brisk, and plantar reflexes were absent due to dystonia. Her score on the Unified Dystonia Rating Scale (UDRS; maximum score, 112) was 81. However, the results of her cognitive and ophthalmic examinations and sensations were all normal.
She underwent a brain magnetic resonance imaging (MRI) at the age of seven, which showed T1 hyperintensities in the basal ganglia, brainstem, cerebellum, white matter bilaterally, and pituitary gland. At the age of 18, MRI scans performed in our hospital demonstrated a greater prominence of T1 hyperintensities (Figure 1, pretreatment). Based on the suspicion of Mn accumulation in the brain, she was tested for serum Mn and the level was elevated at 222.9 µg/L (normal range < 8.0 µg/L). Routine blood tests revealed iron deficiency anemia. Hemoglobin was 9.2 g/dL (normal range is 10.8–13.3 g/dL), serum iron level was 15 µg/dL (normal range is 33–193 µg/dL), total iron-binding capacity was 437 µg/dL (normal range 264–448 µg/dL), and serum ferritin level was 6 ng/mL (normal 13–150 ng/mL). The results of liver function tests and the serum copper, ceruloplasmin, and calcium levels were all normal. The liver ultrasonography findings were unremarkable.

Serial magnetic resonance imaging (MRI) brain scans at pretreatment and 4, 12, and 36 months after chelation therapy. On axial T2-weighted images (axial T2WI, upper row), signal hypointensity in the globus pallidus disappeared 12 months after treatment. After then, the medial globus pallidus shows signal hyperintensity (black arrows). Pretreatment MRI shows hyperintensities on axial T1-weighted images (axial T1WI, middle row) and sagittal T1-weighted images (sagittal T1WI, lower row) in the basal ganglia, brainstem, cerebellum, white matter bilaterally, and pituitary gland. Follow-up MRI scans at 4, 12, and 36 months demonstrate a significant decrease in the previous hyperintensities observed on the T1-weighted images.
Initial analysis of whole exome sequencing (WES) data and Sanger sequencing of SLC30A10 at age 18 was negative. Two years later, a SLC39A14-associated manganism was described by Tuschl et al. [2]. Reanalysis of WES data revealed compound heterozygous mutations in the N-terminal region (c.367C>T, p.Q123*) and the first transmembrane domain (c.512G>A, p.G171E) of the SLC39A14 gene (NM_015359.4, NP_056174.2). This was confirmed by Sanger sequencing. Both mutations have been reported to be pathogenic according to the guidelines by American College of Medical Genetics and Genomics, as listed in ClinVar (rs1554519011, rs1554519303). She inherited one mutated gene each from both her parents. These findings are compatible with the genetic diagnosis of hypermanganesemia with dystonia 2 (HMNDYT2, OMIM ID #617013).
Oral ferrous sulfate was initiated to correct iron deficiency anemia and decrease intestinal Mn absorption. The peripheral iron status was normalized six months after iron supplementation and remained stable (Hb 12.2–13.7 g/dL, serum iron 78–149 µg/dL, TIBC 270–303 µg/dL, serum ferritin 55–75 ng/mL). Chelation therapy with parenteral CaNa2EDTA was started soon after genetic confirmation at the age of 20. The original schedule was to administer 500 mg, twice daily for 5 consecutive days at an interval of 3 weeks. Later, due to inconvenience of the patient, it was continued as a 1-day cycle where 1 g was administered twice, once every week. The chelating agent had no notable side effects during the 5-year period. She was also supplemented with trace minerals, including zinc, to avoid depletion during chelation therapy. Laboratory tests were performed every 3 months to monitor the potential adverse effects of CaNa2EDTA chelation therapy. These included thrombocytopenia, leukopenia, nephrotoxicity, hepatotoxicity, hypocalcemia, and trace metal deficiency [4]. Serum Mn levels gradually decreased, and remained stable at lower levels (Figure 2). Over the past year, her serum Mn levels ranged from 37.5 µg/L to 41.9 µg/L. Brain MRI follow-up at 4, 12, and 36 months after chelation therapy showed a significant decrease in previous hyperintensities on T1-weighted images (Figure 1). T2 hypointensity in the globus pallidus disappeared one year after treatment. Instead, the medial globus pallidus showed signal hyperintensity. However, her dystonia improved insignificantly, with a decrease less than 10% in the UDRS scores. It remained stable for 5 years without further improvement in activities of daily living. Symptomatic therapy with levodopa, trihexyphenidyl, and clonazepam was ineffective.
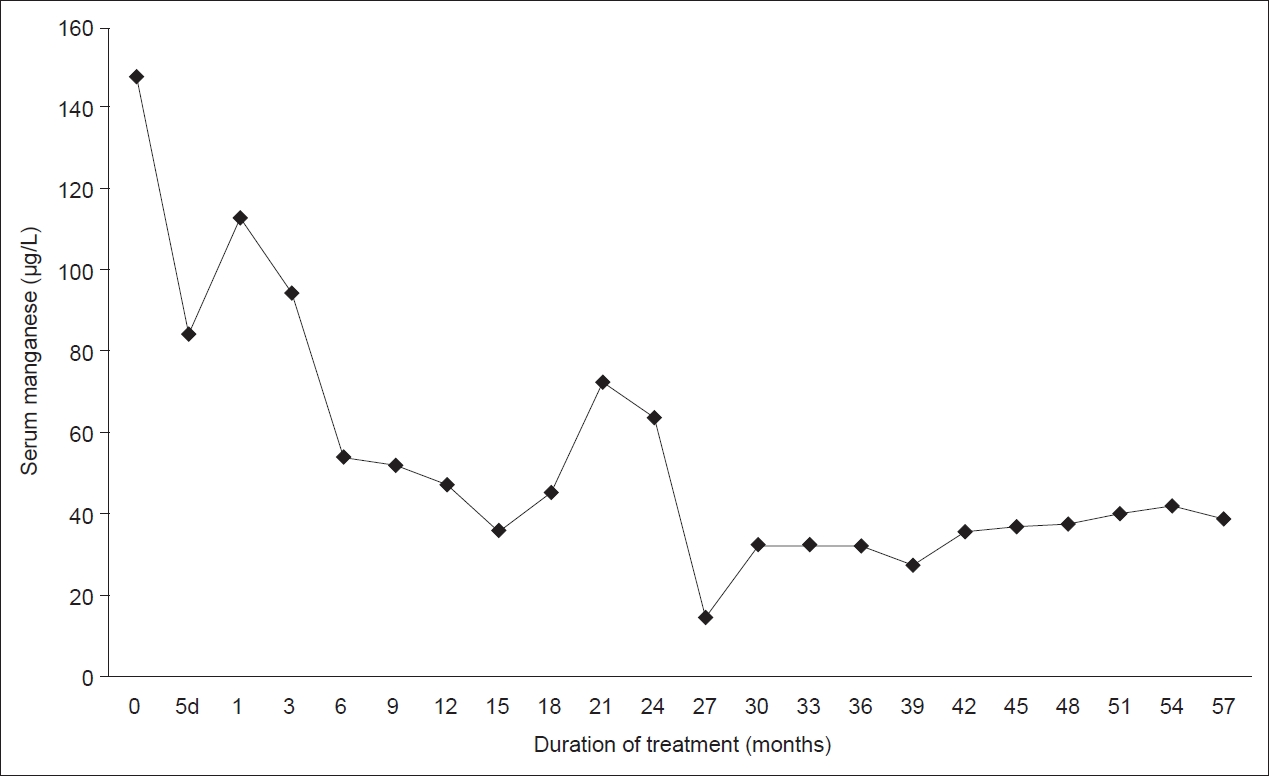
The serial measurements of serum manganese. The graph shows a reduction of serum manganese levels. 5d, 5 days.
DISCUSSION
The clinical and MRI findings of our patient were compatible with SLC39A14-associated manganism [2]. Inherited Mn accumulation disorders caused by SLC39A14 and SLC30A10 mutations typically present with a progressive extrapyramidal phenotype as a consequence of Mn toxicity to the basal ganglia, which is characterized by early onset generalized dystonia-parkinsonism syndrome [5]. White matter involvement can cause spasticity and pyramidal tract signs [1]. Cognition tends to be spared [1,2]. The characteristic MRI brain appearances of cerebral Mn deposition in patients with SLC39A14 mutations are similar to those observed in SLC30A10 deficiency [2]. However, as in our case, individuals with SLC39A14 deficiency do not develop polycythemia and hepatic Mn deposition, unlike those with SLC30A10 deficiency [2,5]. The primary function of SLC39A14 is hepatic Mn uptake, whereas SLC30A10 is a Mn efflux transporter for subsequent biliary excretion [2,5].
Intravenous CaNa2EDTA has been the mainstay of chelation therapy for inherited Mn accumulation disorders [1,3]. In SLC30A10-associated manganism, Mn chelation in combination with iron supplementation leads to improved neurological symptoms with a reduction in T1 hyperintensity and prevents progression of liver disease [1,3]. To date, the results of CaNa2EDTA chelation have been reported only in several patients carrying SLC39A14 mutations [1,2,6-8]. It is even more rare to find long-term follow-up data presented in detail, as has been done in this report.
Efficacy and tolerability can vary according to the patient’s age and disease severity at the initiation of treatment. Existing literature shows that only one 5-year-old patient had marked improvement in neurological symptoms after 6 months of treatment [2]. Two patients with severe disabilities discontinued chelation therapy owing to worsening of motor symptoms [1,2,6]. One patient received an irregular schedule of chelation therapy due to practical limitations and complementary Mn-depleted diets over a 4-year period, starting at the age of five [7]. This did not lead to lower serum Mn levels or a reversal of already established deficits. Another case showed some clinical improvement during the 6-month follow-up period [8]. However, long-term efficacy data are not available. In our case, Mn chelation with intravenous SLC39A14 deficiencyTA effectively lowered blood Mn and reduced T1 hyperintensities on brain MRI. As demonstrated in this report, it may take several months for serum Mn levels to reach sufficiently low levels. Despite these results, our treatment did not significantly improve the motor symptoms. The insufficient response can be partly attributable to T2 hyperintense lesions in the medial globus pallidus, which are compatible with the neuronal loss and reactive astrogliosis that were previously observed in the brain during autopsy [2]. It would have been more effective if the chelation therapy was administered at a younger age and an earlier stage of disease [1,7].
SLC39A14 deficiencyTA was administered intravenously as a 5 day course every 4 weeks in most cases [1]. This dosage schedule was inconvenient for our patient. Hence, later in the course she was switched to a 1-day cycle per week, which is also acceptable. While SLC39A14 deficiencyTA chelation is effective, periodic intravenous administration is burdensome [1]. For convenience, one study administered oral chelators [7], but it proved to be ineffective for Mn excretion. Oral iron supplementation can be beneficial because Mn competes with iron for absorption [1,3]. Moreover, iron supplementation is crucial for patients with iron-deficiency anemia, as in our case. The administration of SLC39A14 deficiencyTA can lead to the reduction of trace metals, such as calcium, zinc, copper, and selenium [6]. Therefore, close monitoring and supplementation of trace elements is required to prevent any deficiencies.
Diagnosis of this inherited Mn accumulation disorder should not be missed because it is potentially treatable. Early detection and treatment may prevent disease progression and even reverse the established deficits.
Notes
Ethics Statement
All procedures in this study involving human participants were performed in accordance with the ethical standards of the Institutional Review Board of the Pusan National University Yangsan Hospital (05-2020-189), Korea, and with the 1975 Declaration of Helsinki and its later amendments or comparable ethical standards. Informed consent was obtained from the patient included in the study.
Conflicts of Interest
The authors have no financial conflicts of interest.
Funding Statement
This was supported by the Basic Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology (2019R1F1A1059873) and a 2022 research grant from Pusan National University Yangsan Hospital. We are grateful to Dr Karin Tuschl for helpful discussions and advice.
Author Contributions
Conceptualization: Jae-Hyeok Lee, Jin-Hong Shin. Data curation: Jae-Hyeok Lee. Supervision: Jin-Hong Shin. Validation: Jae-Hyeok Lee. Visualization: Jae-Hyeok Lee. Writing—original draft: Jae-Hyeok Lee. Writing—review & editing: Jae-Hyeok Lee, Jin-Hong Shin.