Current Status and Future Perspectives on Stem Cell-Based Therapies for Parkinson’s Disease
Article information

Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative disorder after Alzheimer’s disease, affecting 1%–2% of the population over the age of 65. As the population ages, it is anticipated that the burden on society will significantly escalate. Although symptom reduction by currently available pharmacological and/or surgical treatments improves the quality of life of many PD patients, there are no treatments that can slow down, halt, or reverse disease progression. Because the loss of a specific cell type, midbrain dopamine neurons in the substantia nigra, is the main cause of motor dysfunction in PD, it is considered a promising target for cell replacement therapy. Indeed, numerous preclinical and clinical studies using fetal cell transplantation have provided proof of concept that cell replacement therapy may be a viable therapeutic approach for PD. However, the use of human fetal cells remains fraught with controversy due to fundamental ethical, practical, and clinical limitations. Groundbreaking work on human pluripotent stem cells (hPSCs), including human embryonic stem cells and human induced pluripotent stem cells, coupled with extensive basic research in the stem cell field offers promising potential for hPSC-based cell replacement to become a realistic treatment regimen for PD once several major issues can be successfully addressed. In this review, we will discuss the prospects and challenges of hPSC-based cell therapy for PD.
INTRODUCTION
Parkinson’s disease (PD) is one of the most common and complex neurodegenerative disorders, affecting approximately 0.3% of the general population and 1%–2% of the population over the age of 65, and its prevalence will double or triple over the next few decades as the population of the developed world ages [1-3]. The aging of the population imposes a substantial socioeconomic burden, affecting patients and their families’ quality of life and stressing the health care systems supporting them [4-6]. One of the hallmarks of PD is the progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc), leading to dopamine deficiency within the basal ganglia and the appearance of classical parkinsonian motor symptoms (e.g., bradykinesia, tremor, and rigidity) as well as numerous nonmotor symptoms (e.g., depression, constipation, pain, genitourinary problems, and sleep disturbances) [7,8]. Although dopamine-replacement therapy (e.g., L-dopa or dopamine agonists) remains a first-line treatment for PD and greatly benefits many patients, its therapeutic window is limited due to decreasing efficacy and increasing side effects, such as dyskinesias [9,10]. Surgical therapies have proven successful in mitigating some symptoms of PD [11]. Among them, deep brain stimulation (DBS) is commonly used as a nondestructive treatment to normalize motor functions of PD patients by modulating the dysregulated output nuclei of the basal ganglia when pharmacological therapy is exhausted [12]. Despite its effectiveness, there are practical drawbacks to DBS, including higher expenses, risk of infection of the implants, erosion and/or migration of implanted hardware, and surgical replacement of non-rechargeable batteries [13,14]. In addition, there are some lingering questions and concerns associated with possible side effects of DBS, such as deterioration in cognition, speech disturbance, depression, postural instability, and significant body mass gain [15-17]. Recombinant adeno-associated virus (AAV)-based gene therapy is another ongoing nonablative PD therapeutic approach. Recent clinical trials with AAV carrying therapeutic genes, such as aromatic-L-amino acid decarboxylase (AADC), glucosylceramidase or glial cell linederived neurotrophic factor (GDNF), demonstrated the efficacy of this approach for PD as well as for other neurological disorders [18,19]. Nonetheless, together with DBS, these gene therapy strategies are palliative, and their long-term effects remain unclear. Moreover, these therapeutic approaches only offer symptomatic treatments, and until now, none of the available treatments have been able to slow down, stop, or reverse disease progression. Pluripotent stem cells (PSCs) have two distinguishable characteristics, self-renewability and pluripotency, allowing them to give rise to almost any cell type [20]. There are two major types of human PSCs, human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs). Due to their unique and extraordinary features, both hESCs and hiPSCs have been considered potential tools for disease modeling and drug discovery as well as for cell transplantation approaches [21]. Over the past several decades, hPSC-derived functional neurons sufficiently matching the identity of endogenous neurons have provided critical advances for the treatment of PD [22]. In this review, we will discuss the background and recent advances in cell replacement therapy for PD covering the use of fetal tissue, hESCs, and hiPSCs.
PD AS A PROMISING TARGET DISEASE FOR CELL THERAPY
PD is a multifactorial neurodegenerative disorder caused by a complicated interplay of aging, genetics, and environmental factors [23]. The hallmark of PD pathology is a progressive loss of A9-type midbrain dopaminergic (mDA) neurons (mDANs) in the SNpc together with α-synuclein-containing Lewy bodies and Lewy neurites [24,25]. The progressive impairment of the nigrostriatal dopamine pathway resulting from this loss is believed to be responsible for PD’s major motor symptoms. This understanding has led to a new era of cell therapies focusing on replacing the lost dopaminergic innervation with dopamine-producing cells, including ventral mesencephalic dopamine neurons from xenogeneic or aborted human fetal tissues, as a cell source [26-28]. A large body of accumulated data has demonstrated the potential of fetal cell transplantation to offer significant and long-term recovery from PD pathology, providing a “proof-of-concept” for cell replacement therapy (Figure 1) [29-31]. However, ethical and practical concerns regarding tissue availability have limited the widespread use of aborted fetal tissue for transplantation, spurring the need to develop alternative sources of therapeutic dopamine neurons. The availability of hPSCs has led to a resurgence of interest in the concept of cell replacement therapy. In particular, reprogramming technology to convert human somatic cells into induced pluripotent stem cells (iPSCs) allowed the production of desired patient-specific cells with standardized quality and unlimited quantity without ethical concerns [32,33]. Recently, multiple research groups successfully developed direct differentiation protocols to generate functional mDAN populations from hESCs and hiPSCs, leading to successful preclinical outcomes using diverse animal models of PD [34,35]. Based on advancements in our understanding of the molecular mechanisms of mDA neuronal development, this may allow both hESCs and hiPSCs to become standard cell sources of mDA neuronal production for transplantation in PD patients. We will discuss the bench-to-bedside developments of stem cell therapy for treating PD in greater detail below.
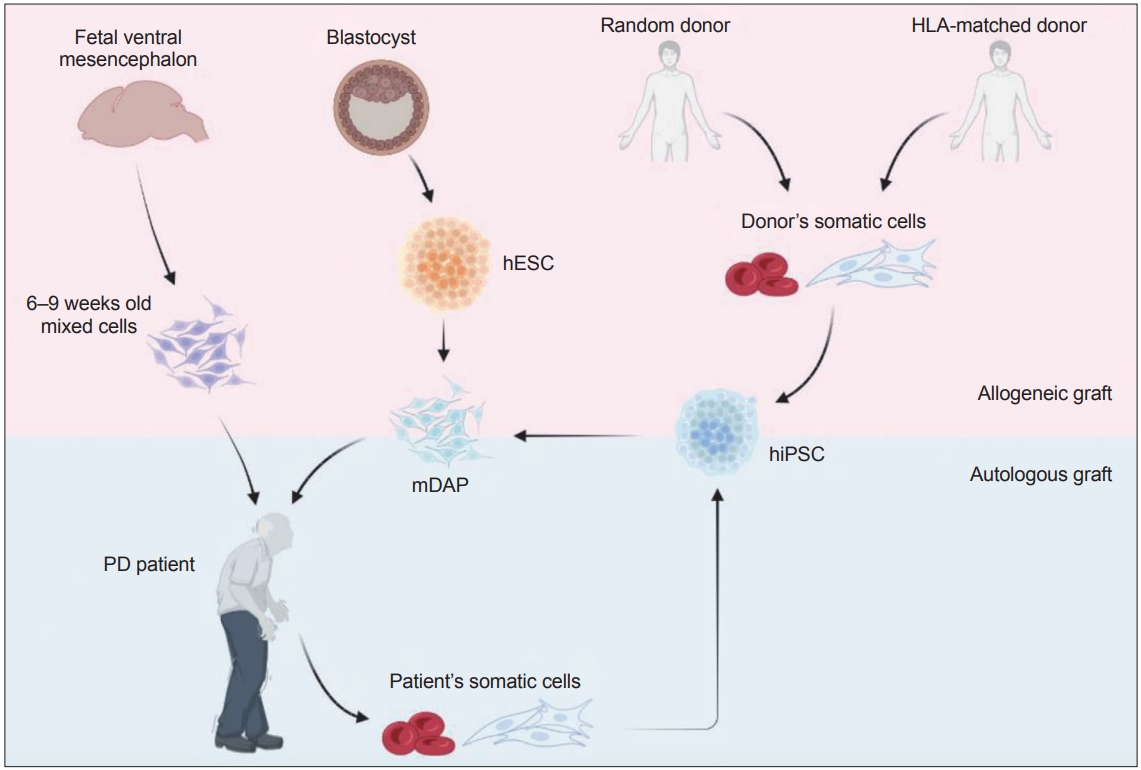
Comparison of potential cell sources for cell replacement therapy for Parkinson’s disease (PD). Transplantation of fetal ventral mesencephalon cells obtained from 6–9 weeks-old aborted human embryos provided proof of concept in support of cell replacement therapy for PD. However, the use of aborted human embryos raised ethical and practical concerns. Human embryonic stem cells (hESC) are an alternative and renewable cell source for producing mDAPs, but suffer from the risk of accumulation of deleterious mutations associated with long-term maintenance and require immunosuppression to prevent graft rejection. Human induced pluripotent stem cells (hiPSC) derived from either random- or HLA-matched donor cells can be selected based on quality assessment (e.g., free from tumorigenicity or toxicity) and used as a universal source for treating PD patients although subject to immunosuppression regimen. While hiPSCs derived from patient’s somatic cells may be an optimal cell source, allowing autologous cell transplantation without any immune rejection issues, it requires additional time and expense. mDAP, mDA progenitor; HLA, human leukocyte antigens.
DEVELOPING CELL THERAPY APPROACHES FOR PD
Mesencephalic fetal cell transplantation as early proof-of-concept studies–lessons and challenges
The early 1980s open-label study by Lindvall et al. [36] first showed that transplantation of human fetal mesencephalic dopaminergic neurons obtained from aborted human embryos at 8–9 weeks gestational age into the striatum of patients with PD could survive in the human brain and resulted in significant improvement of the severe rigidity, bradykinesia, and fluctuations in the patient’s condition. This presumably occurred by restoring striatal dopaminergic transmission, revealing the promise of cell transplantation therapy using fetal tissues [36]. Subsequently, similar open-label studies have been undertaken worldwide and have shown that some patients gained symptomatic relief and restoration of normal dopamine signaling for up to two decades [37-39]. Several follow-up studies showed postmortem evidence of grafted fetal tissue innervating the host striatum with wide outgrowth up to 24 years posttransplantation [40]. However, the results of two NIH-funded double-blind, sham-controlled clinical trials failed to provide statistically significant improvement, and 18 (32%) of 56 transplanted patients even developed adverse effects, mostly graft-induced dyskinesia of unclear etiology [27,31,41,42]. A possible explanation for this motor complication came from in vivo imaging studies of fetal cell-transplanted patients who experienced poor outcomes, showing unregulated dopamine release by cografted serotonergic neurons [27,38,43,44]. Although the original enthusiasm for this approach was dampened by these unsatisfactory clinical outcomes, and a temporal moratorium on fetal cell transplantation was subsequently imposed in the early 2000s, lessons learned from these fetal cell transplantation studies suggested that advancements in cell therapeutic strategies for PD could have the potential to improve patients’ quality of life. In pursuit of this idea, multiple groups in this field launched the “G-Force PD” consortium to collaborate on the optimal development of standardized cell transplantation procedures for clinical application [45]. In line with this, Roger Barker and colleagues initiated a new European trial using human fetal ventral mesencephalon (fVM) tissues, known as TRANSEURO (www.transeuro.org.uk), to develop an efficacious and safe methodology for patients with PD [46]. Although long-term follow-up studies showed that some of the original fetal transplant patients obtained persistent motor symptom relief, demonstrating the efficacy of cell transplantation therapy in the treatment of PD, ethical and practical concerns regarding the use of aborted human fetuses (usually 6–8 fetuses are required per patient) remain hurdles [34,47,48]. In particular, insufficiency of suitable human fetal tissue resulted in delay or denial of many surgical referrals for severe patients with PD in the TRANSEURO study [49]. In addition, multiple variable factors, such as the maturity and purity of fetuses, cell viability between batches, and amounts of grafted cells, highlight the need for new sources of authentic and functional human dopaminergic cells with higher scalability and reliable quality [50].
hESC-derived dopamine cells for allogeneic grafting
The critical lessons learned from the clinical application of fVM cells prompted investigators to explore alternative cell sources, including diverse stem cell types with the capacity to differentiate into multiple cell lineages. A wide range of stem cell types has been identified and extensively characterized in vitro and in vivo. The isolation and establishment of mouse embryonic stem cells (ESCs) in 1981 revolutionized biological research and attracted particular interest due to their indefinite self-renewal and pluripotent differentiation potential, with the capability to generate any cell type of the body [51,52]. Subsequently, Thomson et al. [53] first established hESC lines from human blastocysts, and this new scalable cell source as a replacement for fetal tissue prompted a new era of translational research toward clinical application [54]. Given the expectation that mouse and human mDANs follow largely similar developmental steps and respond to similar factors, translation of the knowledge gained from mouse ESC differentiation studies to human ESCs led to rapid success in preclinical studies using PD xenograft models [34,55]. Earlier studies mainly relied on the use of dopamine-specific morphogens and growth factors (e.g., SHH, FGF8, BDNF, GDNF, and dbcAMP) and coculture methods using mouse stromal cell lines, including PA6 and immortalized human fetal midbrain astrocytes, to induce mDANs from hESCs, but this approach was hampered by a high level of variation within and between studies due to poorly defined culture conditions [56-58]. With the idea that both WNT1-LMX1A and SHH-FOXA2 autoregulatory loops are crucial for the specification and differentiation of mDANs during embryonic development [59-61]. and that midbrain floor plate cells display characteristics of radial-glial-like progenitors that give rise to mDANs [62,63], Lorenz Studer’s group developed a small molecule-based dual SMAD inhibition protocol in conjunction with dual activation of both WNT and SHH signaling pathways at the early stage of hPSC in vitro differentiation. This model resulted in the midbrain-floor-plate-based induction of functional mDANs with therapeutic potential [64,65]. Some representative protocols are shown in Figure 2. These protocols are capable of producing tyrosine hydroxylase (TH)-positive mDANs in vitro, as evidenced by their expression of FOXA2, LMX1A, PITX3, NURR1, EN-1, and the dopamine transporter. Moreover, it is noteworthy that multiple studies by different research groups demonstrated that hESC-derived mDANs displaying A9 characteristics repair the damaged nigrostriatal circuitry and thus restore motor deficits in animal models of PD [66-68]. At the time of writing, there were four registered clinical trials using hESC-derived dopamine cells or human parthenogenetic neural stem cells as the allogeneic cell source for transplantation therapy (Table 1) [47,69-71]. Among these, Wang et al. [70] launched the first clinical trial using hESCs in 2017 (NCT03119636). Based on recent preclinical animal studies, Studer’s group in the US developed a phase 1 clinical trial to determine the safety and efficacy of allogeneic WA09 hESC-derived mDA progenitors (mDAPs) (MSKDA01) for treating advanced PD patients (NCT04802733) [71,72]. In 2017, Malin Parmar’s group in Sweden have also planned to initiate a European clinical trial (the STEM-PD trial) using allogeneic mDAPs derived from human ESCs (EudraCT 2021-001366-38) [47]. Despite the potential benefit of using hESCs for cell replacement therapy, these hESC-derived cells together with fetal tissues are allogeneic and require immunosuppression to avoid graft rejection. Due to the increased risk of infections and side effects, such as the development of malignancies, the condition and duration of immunosuppression remain controversial, and the potential harmful effects are to be defined [73,74].
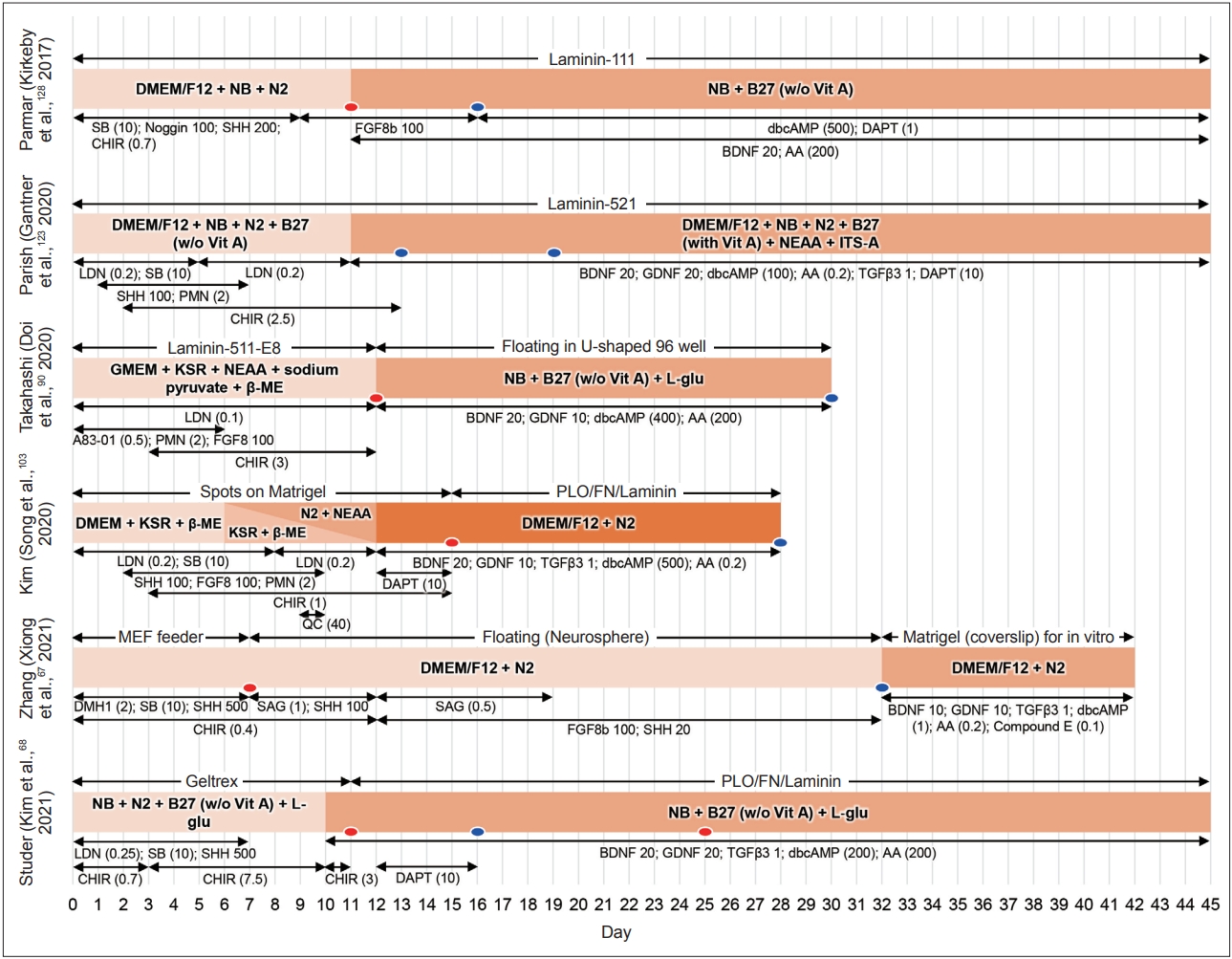
Schematic diagrams of representative differentiation protocols to generate mDA progenitors (mDAPs). Despite significant differences in specific chemicals, their concentrations, culture media, and supplements, all these protocols aim to activate both the WNT1-LMX1A and SHH-FOXA2 autoregulatory loops and inhibit dual SMAD signaling (regulated by BMP and/or TGFβ). Numbers represent concentrations in ng/mL, and those in parentheses show μM. The red and blue ovals represent time points for dissociation and transplantation steps of cell products, respectively. NB, neurobasal medium; SB, SB431542; SHH, sonic hedgehog; CHIR, CHIR99021; FGF8, fibroblast growth factor 8; dbcAMP, dibutyryl cyclic adenosine monophosphate; BDNF, brain-derived neurotrophic factor; AA, ascorbic acid; NEAA, non-essential amino acid; ITS-A, insulin-transferrin-selenium-sodium pyruvate; LDN, LDN193189; PMN, purmorphamine; GDNF, glial cell line-derived neurotrophic factor; TGFβ3, transforming growth factor beta 3; KSR, knockout serum replacement; β-ME, β-mercaptoethanol; L-glu, L-glutamine; DMH1, dorsomorphin homolog 1; QC, quercetin; SAG, smoothened agonist; PLO, poly-L-ornithine; FN, fibronectin; MEF, mouse embryonic fibroblast.

List of clinical trials using fetal tissue or pluripotent stem cell-derived mDA cells
hiPSC-derived dopamine cells for autologous/allogeneic grafting
In 2006, Takahashi and Yamanaka first reported their groundbreaking work showing that they could revert terminally differentiated mouse somatic cells into an ESC-like state, henceforth referred to as “induced pluripotent stem cells” [75]. Soon after this study, Yamanaka’s group and two other groups successfully generated hiPSCs from human somatic cells using the same or similar sets of reprogramming factors [76-78], offering the unprecedented possibility of generating patient-specific hiPSCs free of ethical concerns associated with the destruction of human embryos to obtain human ESCs. The advent of hiPSC technology has ushered an exciting new era for the fields of stem cell biology and regenerative medicine and also offers a useful platform for human ‘disease in a dish’ models and drug discovery [79-81]. Given that the use of primary cells present in pathological conditions is highly limited due to insufficient expandable cellular sources, particularly hard-to-access cells such as brain cells and heart cells from patients, hiPSCs serve as an attractive cell source for disease modeling since they can be derived from easily accessible cell sources, such as skin fibroblasts, blood cells and urinary tract cells [33]. In addition, iPSC technology paved the way for patient-specific hiPSCs to generate target cells that are genetically identical to the graft recipient, allowing for autologous cell therapies for intractable diseases and injuries by evading immune-mediated rejection [82]. Furthermore, research in nonhuman primates demonstrated that mDANs derived from autologous iPSCs elicit a minimal immune response in the host brain compared to those from allogeneic iPSCs [83]. Despite its short history, the therapeutic benefits of using autologous hiPSC-based personalized cell therapy are apparent. The first clinical trial to evaluate the potential of autologous hiPSC-derived cells was initiated by a Japanese group in 2014 [84]. Mandai et al. [84] first performed transplantation of retinal pigment epithelium (RPE) derived from autologous hiPSCs into a patient with advanced neovascular age-related macular degeneration under the retina, and the treatment was reported to improve the patient’s vision, proving the potential of autologous cell therapy. At the time of writing, 3 clinical trials have been conducted using hiPSC-derived cells to treat patients with PD (Table 1). Among these, a pioneering case report, Schweitzer et al. [85] recently presented a milestone in autologous cell replacement therapy, as they showed the feasibility of autologous transplantation of mDAPs derived from a PD patient-specific hiPSC (IND17145) [86-88]. Moreover, another clinical trial testing the efficacy of the administration of autologous neural stem cells to patients with PD was initiated in 2019 (NCT03815071). However, the time-consuming laborious process and high cost for establishing and characterizing patient-specific hiPSCs are considered big hurdles that should be addressed for personalized autologous cell replacement therapy. It is estimated to cost approximately $800,000 to treat one patient with an autologous iPSC-derived cell product [89]. One possibility to overcome these limitations is the use of allogeneic iPSC-based “off-the-shelf” cell therapies. These human leukocyte antigens (HLA)-matched cell products can be established from healthy donors sharing the most common HLA types and thus can target large patient populations, which will significantly reduce the time and cost of each treatment [90]. In addition, cryopreservation enables rigorous preclinical studies to establish batch-specific efficacy of hiPSC-derived cell products that are to be transplanted into humans [91,92]. Furthermore, critical safety issues, including tumorigenic potential, off-target effects, and compatibility with surgical delivery devices, can be addressed through evaluation of the quality of cryopreserved mDAPs before clinical trials [93]. In line with this, Takahashi [94] in Japan initiated a single-arm, nonrandomized, open label phase 1/2 study to evaluate the safety and efficacy of allogeneic hiPSC-derived cryopreserved mDAPs (with variable degrees of HLA matching to the hosts) for the treatment of seven patients with PD in 2018 (Kyoto trial) (UMIN000033564) [94]. However, these allogeneic cell transplantations require patients to receive immunosuppressant medications to prevent possible immune rejection by the recipients.
CRITICAL FACTORS TO BE CONSIDERED FOR CLINICAL USE OF hPSC-DERIVED mDAPs
Advances in our understanding of human PSC research have allowed the establishment of a greater pool of hiPSC lines and NIH-registered hESC lines with diverse genetic backgrounds that can be easily accessed and distributed to researchers, helping to accelerate the development of therapies across disease areas [95]. Currently, PSC-derived mDAP transplantation is a widely accepted therapeutic modality to restore motor and nonmotor functions in patients with damaged dopamine systems [93]. However, despite efforts to establish general guidelines for mDA neuronal differentiation of hPSCs, the concerning lack of reproducibility and consistency among different laboratories has restricted the potential of hPSC-based cell therapies for PD to restore the function of mDANs [96,97]. To fully realize the potential of hPSC-based cell therapy for PD, both the safety and efficacy of transplanted cells should be consistent. There are some critical issues for the application of hPSC-derived mDAPs as universal therapeutic approaches for treating patients with PD, as described below:
Autologous stem cells vs. allogeneic stem cells as initial cell sources
In general, all cell products intended for clinical use as a long-term treatment for patients with PD require a level of safety much greater than that for cells used for developmental and mechanistic studies. The safety of the cell sources for transplantation should be principally ensured by avoiding the use of potentially harmful viral agents and genetic materials that can integrate or disrupt host physiology. The first step for cell transplantation therapy is identifying and choosing a good source of stem cells, which requires considering a number of factors, including the genomic integrity of hPSCs [98]. The use of well-characterized hESC lines, such as the WA09 hESC line, may be good for the purpose of allogeneic cell transplantation since those cell lines have been used and well characterized by numerous researchers for both basic and preclinical studies over two decades. Nevertheless, given that PSCs accumulate 3.5 ± 0.5 base mutations per genome per population doubling, prolonged in vitro culture of these hESC lines can result in genetic aberrations, including recurrent chromosomal abnormalities and point mutations in cancer-related genes found in human malignant tumors [99-101]. Specifically, TP53, a well-known tumor suppressor gene, is the most mutated gene in WA09 hESCs, with four individual mutations (P151S, R181H, R248Q, and R267W), conferring a strong selective growth advantage to mutant cells [102]. Compared to hESCs, patient-derived hiPSCs at early passages are free from any somatic mutation issues caused by prolonged in vitro culturing and thus are suitable for mDAP generation for autologous cell therapy for PD patients [103]. In addition, of the nonintegrating reprogramming methods, the use of synthetic mRNA or Sendai virus to date represents the most advanced approaches allowing the generation of clinical-grade hiPSCs more efficiently at earlier passage numbers (< 10 passages) from patients’ own cells, thus accelerating the production of suitable derivatives [104-106]. Furthermore, using a patient’s own hiPSCs for autologous stem cell transplantation eliminates the risk of tissue rejection [107]. This has been verified by Su-Chun Zhang and colleagues, who transplanted autologous iPSC-derived mDAPs back into the brains of donor parkinsonian monkeys. They reported that monkeys receiving autologous mDAPs, but not allogeneic mDAPs, showed significant long-term improvement in both motor and nonmotor functions without immunosuppression, demonstrating the feasibility of autologous cell therapy for PD [108,109]. However, for autologous iPSC-based cell therapy, there are still key issues that should be considered. Among them, the transplantation of iPSC-derived mDANs could predispose the patient to further neurodegeneration if the cells retain the intrinsic genetic defects giving rise to the condition [110]. Although genome editing techniques, such as zinc finger nucleases, transcription activator-like effector nucleases or clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated 9 systems, are available to correct patients’ mutated genes, off-target effects and mosaic mutations remain hurdles that need to be addressed before this approach reaches clinical application [111]. Moreover, the time- and cost-intensive efforts in generating iPSCs from donor cells and differentiating them into functional mDANs for transplantation are other drawbacks of autologous cell therapy. To overcome these limitations, there have been continuous efforts to utilize HLA-matched, allogeneic hiPSCs as a universal source to reduce immunogenicity risks in recipients with a minimal requirement for immunosuppression [112-114]. For instance, Taylor et al. [112] established an iPSC bank from 150 selected homozygous HLA-typed donors that matched approximately 93% of the United Kingdom population with a minimal requirement for immunosuppression [112]. Shinya Yamanaka’s and Jun Takahashi’s groups are also conducting an iPS cell stock project to establish an allogeneic iPSC bank covering approximately 90% of Japan’s population for treatment purposes [115,116].
Derivation of proper dopamine cell type for transplantation
Selective generation of specific mDA neuronal populations from hPSCs is another issue to be considered for clinical application of hPSC derivatives for patients with PD. Based on their molecular and electrophysiological properties, mDANs have been categorized into three distinct anatomical clusters: the retrorubral field (RrF, also known as the A8 group), the SNpc (also known as the A9 group), and the ventral tegmental area (VTA, also known as the A10 group). Approximately 200,000–420,000 TH+ dopamine neurons reside bilaterally in the SNpc of adult humans, and 60,000–65,000 TH+ dopamine neurons reside bilaterally in the VTA [117,118]. Of these subtypes, the A9 SNpc dopamine neurons are mostly affected and are responsible for many PD motor features, while other subtypes of neurons are less affected. Although several variant protocols for inducing mDANs have been developed from the concept of dual SMAD inhibition that activates both WNT and SHH signaling pathways to prevent the differentiation of hPSCs into meso/endodermal lineages, these methods are still not completely free from drawbacks arising from contamination with other neural types, including GABAergic, cholinergic and serotonergic neurons that are not relevant in PD [119,120]. The presence of large numbers of serotonergic neurons as part of heterogeneous cell populations to be transplanted can cause adverse side effects, such as graft-induced dyskinesia [43]. Therefore, a key question is how we can generate homogenic A9-type dopamine neurons from hPSCs with full potential to reinnervate the host brain and rescue motor and nonmotor deficits in recipients. Many recent efforts have been devoted to optimizing mDA neuronal differentiation from hPSCs by optimizing culture conditions [34]. For instance, diverse studies have explored different concentrations of CHIR99021 (CHIR), an activator of WNT signaling, to induce mDANs. An initial study by Kriks et al. [65] suggested that 3 μM CHIR induces mDANs, while Kirkeby et al. [121] and Xi et al. [122] reported that 0.4–0.7 μΜ CHIR was optimal for the specification of mDANs and that a higher CHIR concentration (> 1 μM) promoted differentiation into hindbrain neurons. In contrast, Gantner et al. [123] recently showed that lower CHIR concentrations (<1.0 μM) produced OTX2 and FOXA2 double-positive forebrain dopamine neurons, while higher concentrations of CHIR (2.5 μM) efficiently produced mDANs. In a more recent study, Studer’s group reported that the optimal concentration of CHIR for inducing mDANs may vary based on media components used in the differentiation process and that biphasic activation of the WNT signaling pathway by treating cells with different CHIR concentrations (initially 0.7 μM between Days 0 and 4; 7.5 μM between Days 4 and 10; 3 μM from Day 10) can improve mDA neuronal derivation [68]. Moreover, collaboration studies between Jun Takahashi’s and Parmar’s groups adopted a flow cytometry-based sorting technique to obtain CORIN-positive mDAPs with enhanced purity from a heterogeneous population of in vitro differentiated cells and demonstrated significant motor recovery without dyskinesia or tumor formation after transplantation in PD animal models [124,125]. However, despite this progress, there are still huge variations in efficacy and adverse effects seen with grafted cells; thus, there is demand for developing a fully optimized and standardized procedure for testing the functional efficacy of the resulting mDAPs for broader application. In addition, as most current in vitro protocols produce approximately equal ratios of A9- and A10-type mDANs from hPSCs, it is unclear whether improved purity of A9-type mDANs would improve clinical outcomes in transplanted PD patients. Consequently, the proper concentrations of CHIR and other neurotrophic factors (e.g., BDNF and GDNF) should be optimized for individual culture conditions to generate authentic mDAPs. Another critical factor for the clinical application of hPSC-derived mDAPs is the identification of the ideal cell stage of differentiation to be used. As progressive loss of mDANs in the SNpc is responsible for PD pathogenesis, it is logical to transplant fully differentiated mDANs to replace those that are lost. Several groups have shown the feasibility of this approach: Studer’s group reported that human ESC-derived mDANs were efficiently engrafted in mouse, rat, and rhesus monkey models of PD [65]. Ole Isacson and colleagues have shown that unilateral engraftment of autologous iPSC-derived dopamine neurons on Day 49 results in a gradual onset of functional motor improvement and increased motor activity in a nonhuman primate model of PD [126]. However, a study by Chung demonstrated that Otx2+ Corin+ floorplate progenitors were also capable of generating functional TH+ dopamine neurons in vivo following transplantation into the mouse striatum and significantly improved behavioral dysfunction in 6-OHDA-lesioned mouse models [127]. Furthermore, since transplantation of early-stage cells can cause teratoma formation, while later-stage cells exhibit poor survival outcomes in the host brain [34], it is imperative to determine the ideal cell stage among floorplate progenitors, mDAPs, and fully differentiated mDANs. As shown in Figure 2, recent representative studies have tested the efficacy of mDAPs following in vitro differentiation of hPSCs for 16–32 days when the cells express conventional mDAP markers, such as FOXA2, LMX1A, CORIN or OTX2. Both Studer’s group [68], and Parmar’s group [91,128] showed the in vivo efficacy of differentiating mDAPs to Day 16 despite their different culture conditions, whereas Su-Chun Zhang’s group [108] produced mDAPs as floating cell aggregates until transplantation at Day 32 of differentiation to determine the in vivo efficacy of grafted cells in host brains. A recent study by Hiller et al. [129] demonstrated a superior efficacy of Day 17 mDAPs compared with Day 24 immature neurons or Day 37 postmitotic neurons in 6-OHDA-treated rats, as evidenced by the numbers of surviving neurons, innervation, and functional recovery. Therefore, broad comparative analysis of different media compositions (e.g., basal media, supplements, morphogens, small molecules, and their concentrations and timing) across these protocols should be thoroughly conducted to advance our understanding of mDA neuronal development and establish a “gold standard” protocol for the induction of clinically applicable mDAPs with optimal therapeutic potentials for PD.
Optimal cell number
Determining the optimal cell number to be implanted is an additional critical factor that should be considered for cell transplantation therapy for PD because this may vary depending on cell type and viability, administration route, and degree of symptomatic relief. Several preclinical studies have shown functional recovery in animal models of PD with transplanted mDANs using different conditions (Table 2) [47,65,70,71,90,92,103,125,130-137]. The lesson learned from various previous studies using dopamine neurons derived from human fetal tissues or PSCs is that even a single round of transplantation could be sufficient for long-term survival and functional outcomes [138]. Although there is no current consensus on the optimal number of cells for treating humans, based on the findings from human fetal cell transplantation studies, approximately 40,000–80,000 TH+ mDANs with a unidirectional axonal architecture of ≥ 3–5 cm in length and dopamine release of ≥ 7 ng/mg of tissue may be required to achieve meaningful clinical improvements in PD patients [139]. Since the majority of grafted dopamine neurons die within a week posttransplantation [140], to compensate for this severe loss and provide an adequate number of functional dopamine neurons capable of replacing damaged cells in the brain, transplantation of an excess number of cells appears to be needed. In line with this idea, a first case report of autologous hiPSC-based therapy for a single patient with PD demonstrated that injection of 4 million mDAPs derived from autologous hiPSCs into the striatum of a patient with progressive idiopathic PD restored the deteriorated motor functions without any adverse events related to the intervention at 18–24 months after implantation [85]. In addition, in the ongoing phase 1/2 clinical trial (UMIN000033564), Takahashi et al. [94] used approximately 5 million cells per patient, split into three tracts per side, and each tract was divided into 4 delivery points receiving 200,000 cells for a total of 12 points per side. In addition to optimizing the total cell number to be delivered, an optimal concentration of cells should be determined since, at high concentrations (≥ 100,000 cells/μL), the solution becomes viscous and may cause needle clogging and uneven injection flow or lead to low cell survival due to limited oxygen and nutrient diffusion [141]. Moreover, a maximum cell concentration within a certain volume can also vary widely with respect to different cell sizes. In terms of continuous efforts to enhance the survival and stable integration of grafted mDAPs, several studies have been proposed to administer recombinant growth factors, including GDNF, neurturin (NTN), and platelet-derived growth factor. For instance, a recent study by Gantner et al. [136] demonstrated that transgenic overexpression of GDNF within the host brain significantly promoted the maturation, plasticity, and functional integration of grafted cells in a rodent model of PD, demonstrating the potential of neurotrophic gene therapy strategies to improve outcomes of grafted hPSC derivatives.

List of preclinical transplantation studies of hESC- or hiPSC-derived mDA cells
Safety of hPSC-derived cell grafts
The potential tumorigenicity risk from undifferentiated PSCs among transplanted cells is one of the major concerns associated with translational stem cell therapy [142]. Somewhat surprisingly, even in the first clinical trial of autologous RPE cells by Mandai et al. [84], residual undifferentiated hiPSCs that may retain the potential for teratoma formation were detected in transplanted patients [84]. Therefore, it is imperative to establish safe and effective mechanisms to detect any residual undifferentiated cells with tumorigenic potential and remove those cells from the transplantable cell population. A fluorescence-activated cell sorting strategy using antibodies against specific undifferentiated hPSCs or authentic mDAP surface markers is a powerful option to obtain a pure population of transplantable cells. As mentioned above, both Jun Takahashi’s and Malin Parmar’s groups adopted a surface marker-based cell purification step to enhance the purity of cell products and reduce the risk of tumor formation before grafting [124,125]. Furthermore, other efforts were made to completely avoid any possible contamination with undifferentiated cells. Notably, two groups have identified small molecules that can selectively eliminate undifferentiated hPSCs without interfering with differentiation capabilities [143,144]. Ben-David et al. [143] identified 15 pluripotent cell-specific inhibitors through a high-throughput screen of over 52,000 small molecules and further identified an inhibitor of stearoyl-CoA desaturase as the most efficient small molecule for preventing teratoma formation from tumorigenic undifferentiated cells. In addition, using a different strategy, Lee et al. [144] reported that two anti-apoptotic genes, BIRC5 and BCL10, are highly enriched in hPSCs and that quercetin and YM155 can selectively eliminate undifferentiated hPSCs by inhibiting the expression of BIRC5, suggesting a potential small molecule-based strategy for controlling the safety of stem cell therapy for PD. Moreover, a more recent study by de Luzy et al. [145] demonstrated the feasibility of a suicide gene-based system (e.g., thymidine kinase linked to cyclinD1) to selectively ablate proliferating undifferentiated cells, leading to improved safety and purity of hPSC-derived grafts in a rat model of parkinsonism. However, the potential risk of silencing/mutation of the suicide gene or loss of heterozygosity in donor cells can limit the ability of this suicide-based technology; thus, further study will be required to improve the use of this suicide system with context-dependent attention.
Immune response to grafted cells
The possibility of grafted-cell/tissue rejection by the host immune response is another major obstacle and concern in stem cell-based transplantation for PD. As briefly mentioned above, the host immune response is one of the major hurdles impacting allogeneic cell transplantation, which is fundamentally different from autologous transplantation. Although the brain has been considered an “immune-privileged” organ, in a humanized mouse model, immune rejections to HLA-mismatched mDAP (C4-hu+H9-mDAP, K1-hu+C4-mDAP) grafts have been reported [85]. Since the integrity of the blood–brain barrier is damaged by the surgical procedure, it compromises the immuneprivileged status of the host brain and potentially triggers the entry of immune cells, thus requiring immunosuppressive regimens to prevent graft rejection and promote cell survival and innervation [93,146]. Although somatic cell nuclear transfer-derived ESCs (SCNT-ESCs), as an autologous cell source for cell therapy, may constitute an attractive alternative to overcome the limitation associated with allogeneic ESC-derived cells, this approach is also not entirely free from ethical and immunogenic limitations because SCNT-ESCs acquire mitochondria from the oocyte donor, which may induce alloimmunity after transplantation [147]. One possible solution to avoid or reduce the immune rejection issue of grafted cells is to generate hiPSC lines from common HLA-homozygous donors [148]. Since HLA plays a key role in self/nonself-recognition and discrimination against foreign antigens, the HLA-matching hiPSC-based approach allows the immune system of the recipient to recognize the transplanted cells as a part of its own body and minimizes the risk of tissue rejection postimplantation [149]. In line with this, there are rising attempts to establish hPSC banks worldwide. As mentioned above, Yamanaka’s and Jun Takahashi’s groups estimated that hiPSC lines derived from approximately 140 unique HLA-homozygous donors would be sufficient to cover up to 90% (up to 160,000 individuals) of the Japanese population [115,116], assuming allografts using HLA-homozygous iPSCs as a therapeutic alternative to autologous grafts. Based on genetically diverse populations in the United States, large-scale hPSC banks are currently being established to cover a large percentage of the population, including European Americans, African Americans, Hispanics, and Asians [150]. Despite these efforts, the immune response that occurs even with HLA-matched cells is probably due to an indirect pathway caused by H-Y minor histocompatibility antigens or innate immunity caused by natural killer cells [113,151]. Likewise, it is possible that an immune response may occur if immunogenic antigens such as Zg16 and Hormad1 are abnormally overexpressed despite autologous cell sources [152,153]. Even if these are not the major parts, they should not be ignored. In addition, due to a potential burden of immunosuppression on graft recipients, the administration of immunosuppressants needs to be balanced with the predicted benefits of cell transplantation therapy. Therefore, further evaluation of immunosuppressive regimens should include optimization of the type, dose, and duration of immunosuppressants to avoid variations between different individuals and to decipher the safety and efficacy for treating patients with PD.
Patient stratification for transplantation
Successful clinical outcomes of cell replacement therapy depend on the nigrostriatal dopaminergic system integrity of the recipient. As appropriate reinnervation of the grafts into the host striatum may play a critical role in functional recovery from PD, a sufficiently functional nigrostriatal system should be present in recipients before transplantation. An early study by Anders Bjorklund and colleagues reported that the functional efficacy of intrastriatal transplants of fVM cells greatly depended on the severity of damage to the host nigrostriatal system [154]. Functional recovery seen in rats with > 70% dopamine denervation was superior to that in rats with complete lesions of the mesencephalic dopamine system, indicating that spared portions of the host dopamine system may be necessary for the grafts to exert their optimal functional effect [154]. In addition, a previous double-blind, placebo-controlled study reported promising results showing a beneficial effect of fVM tissue transplantation in younger PD patients but not older patients [31,155]. These results raised critical questions: Which patients should be selected for cell therapy, and what level of dopaminergic degeneration warrants neurorestorative intervention using cell therapy? Indeed, current cellular therapeutic strategies for treating PD aim to choose individuals with earlier stages or less severe symptoms based on their disease progression profiles and pathology severities [38]. The TRANSEURO program therefore selectively put younger and early-stage PD patients into an observational cohort study with long-term clinical follow-up assessment [49]. In addition, the program also provides substantial recommendations, including patient-selection criteria and standard operation procedures, useful for stem cell-based future cell replacement therapies [49].
FUTURE DIRECTIONS
Application of hypoimmunogenic PSC-derived mDANs
Considering that the success of PSC-derived cell therapy for PD depends on immune matching between the implanted mDANs and the host patient, there is no doubt that autologous hiPSCs offer an ideal and unlimited cell source that can be used for patient-specific treatments without immune rejection concerns. However, despite its usefulness, the processes of generating and characterizing autologous hiPSCs from patient-specific somatic cells and expanding and characterizing those hiPSCs in sufficient numbers for therapeutic use are time-consuming, laborious, and costly. Furthermore, these autologous cells are only usable to treat donor patients, which is an obstacle to the widespread application of autologous hiPSCs for personalized cell therapies. Although HLA-matching hESC or hiPSC lines are considered an alternative source to reduce the immune rejection associated with allogeneic transplantation, this strategy may be less practical and effective with ethnically diverse populations, and it remains unclear whether its application completely obviates the need for strong immunosuppressant medications in recipients, as mentioned above [156]. Recent advances in genome editing technologies, including the CRISPR–Cas system, have led to new revolutions and innovations in the field of cell-based therapies [157]. In particular, CRISPR–Cas-based genome editing has been instrumental in the development of ‘off-the-shelf’ engineered PSCs for use as therapeutics. An important milestone study by Sonja Schrepfer and colleagues demonstrated that genomic knockout of major histocompatibility complex (MHC) class I and II genes (β2-microtubulin and chromatin-modifying enzymes with class II transactivator, respectively) and simultaneous overexpression of the transmembrane protein CD47 minimizes the immunogenicity of both mouse and human iPSCs. Furthermore, endothelial cells, smooth muscle cells, and cardiomyocytes derived from these hypoimmunogenic iPSCs evade immune rejection in fully MHC-mismatched allogeneic recipients without the use of immunosuppression, thus offering proof of concept that universal donor iPSCs for stem cell-based cell therapies for patients in need of replacing damaged tissues with functional engineered counterparts are a viable option [158,159]. Akitsu Hotta and colleagues also developed tailored HLA-editing strategies using CRISPR–Cas9 to establish pseudohomozygous iPSC lines by disrupting HLA-A and HLA-B biallelically but retaining a single HLA-C allele that enables iPSC derivatives to evade the inhibitory and destructive activities of CD8+ T and NK cells [160,161]. Although these gene editing-based approaches can potentially enable the large-scale production of hypoimmunogenic tissues from allogeneic healthy donor hPSCs that could be ready to treat patients with different histocompatibility, they are still in the early development stage, and several issues need to be addressed before these hypoimmunogenic hPSCs become more widely used for cell transplantation therapies. A major concern of this approach is that it is unclear whether major histocompatibility antigen-manipulated cells would remain hypoimmunogenic or immune privileged in human hosts posttransplantation [159]. In addition, ectopic expression of CD47 may cause deficiencies in host immune responses and protect the grafted cells from immune-system-mediated rejection if they are converted to the cancerous state [161]. Therefore, further studies are required to investigate the effects of transplantation of hypoimmunogenic hPSC derivatives in allogeneic recipients with long-term observations.
Combined cell and gene therapy
In addition to cell therapy, gene therapy has also been extensively explored as an option for treating PD [162]. While transplantation of mDANs was intended to replace damaged cells and provide dopamine directly to the striatum, gene therapy using viral vectors has mainly focused on the restoration of dopamine synthesis, neuroprotection, and spared endogenous host dopaminergic neural circuitry via delivery of dopamine-synthesizing enzymes (e.g., TH, AADC, and GCH1) or neurotrophic factors (e.g., GDNF and NTN) directly to the striatum as a means to enhance dopamine transmission [163]. In addition, given that recessively inherited forms of PD have been linked to mutations in at least five different genes, including α-synuclein (A30P and A53T), PARKIN, DJ1, PINK1, and LRRK2, with seemingly diverse functions [164], gene therapy with the introduction of either small interfering RNAs against the mutated genes or ectopic expression of related regulatory genes, such as key dopamine biosynthetic enzymes, can minimize the cytotoxicity caused by genetic mutations and exert potent protective effects on dopamine neurons against the pathogenesis of PD. In particular, α-synuclein-positive Lewy bodies and Lewy neurites were detected in grafted neurons in PD patients who received transplants of embryonic mesencephalic tissue more than a decade prior to their deaths, suggesting “host-to-graft disease propagation.” [165,166] Considering the possibility that such pathologies may be toxic to transplanted cells over time and limit the long-term efficacy of cell therapy, a recent study showed an interesting possibility that reducing or completely removing α-synuclein alleles from hPSCs using CRISPR/Cas9n-mediated gene editing may yield pathology-resistant mDANs [167]. Furthermore, gene therapy treatments are based on a single intervention principle, resulting in long-term (almost lifetime period) stability of therapeutic outcomes in the brain, which can reduce the financial burden of treatment to both individuals and society [168]. Although several questions remain, including the choice of the most efficient vector and optimal dosage for gene delivery, there are multiple ongoing efforts to further improve the potential of gene therapy by combining it with other therapeutic approaches [169]. Among them, a more recent study by Clare Parish and colleagues demonstrated that homotopic transplantation of hESC-derived mDANs recapitulates long-distance, anatomically precise innervation of appropriate targets throughout the host brain, including robust striatal innervation when combined with forebrain GDNF delivery, leading to the restoration of motor functions in a rodent model of PD [137]. Despite the fact that controlling the expression of viral vectors to deliver ectopic GDNF levels in vivo has been elusive, this ‘proof-of-principle’ study highlights the promising potential of combining cell and gene therapy to provide an optimal therapeutic intervention for the effective treatment of patients with inherited parkinsonism. Therefore, to realize this potential, a more comprehensive assessment of the long-term impact of this combined therapy is required to ensure its safety and efficacy as a prospective therapeutic candidate for PD.
CONCLUSION
Recent progress in the field of stem cell research offers novel therapeutic options for patients who are refractory to current treatment strategies by providing patient-tailored PSCs that can differentiate into multiple types of cells in vitro and in vivo. Notwithstanding the abovementioned remaining technical and scientific challenges, there are increasing numbers of preclinical and clinical studies using hPSC derivatives for treating human neurodegenerative disorders, including PD [149]. Despite this progress, there are some concerns regarding cell transplantation therapy for PD. For instance, Dr. Ron Alterman recently voiced his criticisms on the first case report describing a patient treated with autologous mDAPs for PD in the following ways: 1) past cell therapy trials have failed despite promising open-label phase 1 studies, 2) the placebo effect may account for some aspects of these failures, 3) side effects such as graft-induced dyskinesia were another cause of trial failure, and 4) not all underlying pathophysiological mechanisms of PD are addressed by cell therapy [170,171]. Although the starting point for these criticisms is that cell therapy will likely provide no more effective symptom control than is currently available with proven therapies, such as DBS and dopamine medications [172], presently this is unknown, and ongoing investigations will help clarify this matter. Nevertheless, a conceptual advantage of stem cell-based therapy is that this approach alleviates parkinsonian symptoms (such as DBS or dopamine pharmacotherapy) as well as restores synapse formation and dopamine turnover between transplanted cells and preserved host neurons and rescues both motor and nonmotor deficits in PD patients. Furthermore, compared to the late 1990s fetal cell transplantation approaches, additional advantages of current PSC-based therapeutic approaches include: 1) novel strategies to reprogram cells (including the use of Nobel Prize-winning approaches for somatic cell reprogramming), 2) the practical ability to utilize PSCs with reduced or no graft rejection, and 3) systemic manufacturing methods to generate clinical-grade mDAPs and ensure their safety and efficacy before transplantation [171]. Although stem cell-based therapies for PD have some limitations (e.g., dopamine-related stem cell therapy may not correct all known nonmotor symptoms of PD resulting from nondopaminergic pathology; none of the cell-based therapies has yet undergone a successful phase 3 clinical trial), and the use of hPSC derivatives may continue to raise concerns about safety and feasibility, intensive scientific efforts have been committed – and are ongoing – to improve and further optimize the hPSC-based cell therapeutic approach to make it safer, more efficient, and less costly. Continuing effort, including both basic and clinical investigations, into a deeper understanding of PD etiology is imperative to translate our discoveries into novel therapeutic avenues to treat both motor and nonmotor PD symptoms. In addition, considerable commercial and governmental investment in this field will also be important for overcoming practical limitations, such as the cost of manufacturing mDAPs to make hPSC-based cell therapy a viable therapeutic approach available to large numbers of PD patients.
Notes
Conflicts of Interest
The authors have no financial conflicts of interest.
Funding Statement
This work was supported by NIH grants NS127391 and OD024622, the Parkinson’s Cell Therapy Research Fund at McLean Hospital and Massachusetts General Hospital, and the Masson Family Endowed Scholar in Neurosurgery, Massachusetts General Hospital.
Author Contributions
Conceptualization: Young Cha, Kwang-Soo Kim. Funding acquisition: Kwang-Soo Kim. Supervision: Kwang-Soo Kim. Writing—original draft: Young Cha, Kwang-Soo Kim. Writing—review & editing: all authors.