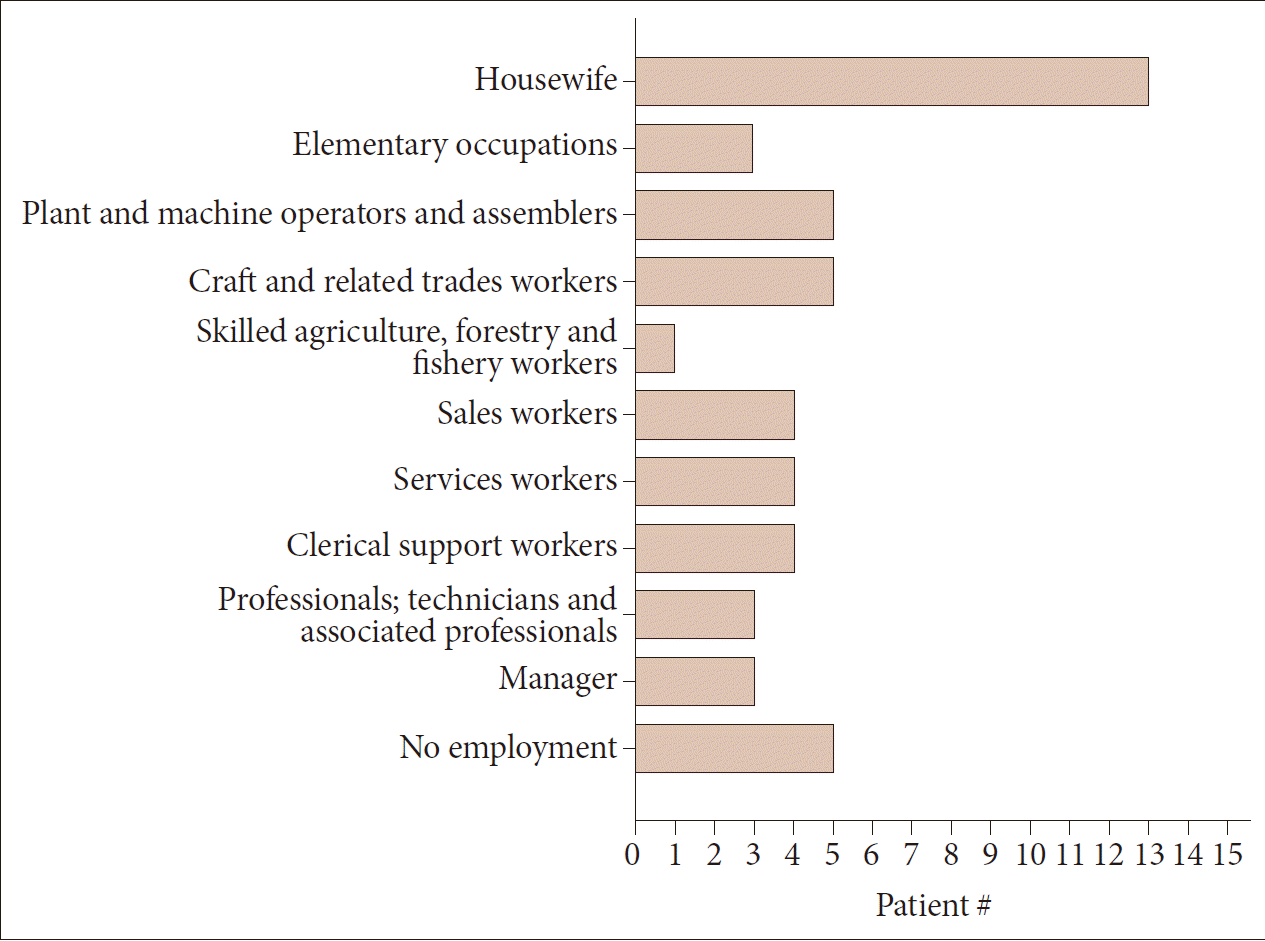E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 8(1); 2015 > Article
-
Original Article
Current Status of Huntington’s Disease in Korea: A Nationwide Survey and National Registry Analysis - Hyun Sook Kim1, Chul Hyoung Lyoo2, Phil Hyu Lee3, Sang Jin Kim4, Mee Young Park5, Hyeo-Il Ma6, Jae Hyeok Lee7, Sook Kun Song8, Jong Sam Baik9, Jin Ho Kim10, Myung Sik Lee2
-
Journal of Movement Disorders 2015;8(1):14-20.
DOI: https://doi.org/10.14802/jmd.14038
Published online: January 13, 2015
1Department of Neurology, CHA Bundang Medical Center, CHA University, Seongnam, Korea
2Department of Neurology, Gangnam Severance Hospital, Yonsei University College of Medicine, Seoul, Korea
3Department of Neurology and Brain Research Institute, Severance Biomedical Science Institute, Yonsei University College of Medicine, Seoul, Korea
4Department of Neurology, Busan Paik Hospital, Inje University College of Medicine, Busan, Korea
5Department of Neurology, Yeungnam University Medical Center, Daegu, Korea
6Department of Neurology, Hallym University Sacred Heart Hospital, Anyang, Korea
7Department of Neurology, Pusan National University Yangsan Hospital, Yangsan, Korea
8Department of Neurology, Jeju National University Hospital, Jeju, Korea
9Department of Neurology, Sanggye Paik Hospital, Inje University College of Medicine, Seoul, Korea
10Department of Neurology, Chosun University School of Medicine, Gwangju, Korea
- Corresponding author: Hyun Sook Kim, MD, PhD, Department of Neurology, CHA Bundang Medical Center, CHA University, 59 Yatap-ro, Bundanggu, Seongnam 463-712, Korea/Tel: +82-31-780-5480/Fax: +82-31-780-5269/E-mail: hskim626@cha.ac.kr
Copyright © 2015 The Korean Movement Disorder Society
ABSTRACT
-
Objective
- Huntington’s disease (HD) is a rare neurological disorder, and its current status in Korea is not well investigated. This study aims to determine the prevalence and incidence of HD and to investigate the clinical features of HD patients in Korea.
-
Methods
- We estimated the crude prevalence and annual incidence of HD based on the databases of the Rare Diseases Registry (RDR) and the National Health Insurance (NHI). The clinical data of genetically confirmed HD patients was collected from 10 referral hospitals and analyzed.
-
Results
- The mean calculated annual incidence was 0.06 cases per 100,000 persons, and the mean calculated prevalence was 0.38 based on the NHI database. The estimated crude prevalence based on the RDR was 0.41. Of the sixty-eight HD patients recruited, the mean age of onset was 44.16 ± 14.08 years and chorea was most frequently reported as the initial symptom and chief complaint. The mean CAG repeat number of the expanded allele was 44.7 ± 4.8 and correlated inversely with the age of onset (p < 0.001). About two-thirds of the patients have a positive family history, and HD patients without positive family history showed a delay in onset of initial symptoms, a prolonged interval between initial symptom onset and genetic diagnosis and a delay in the age of genetic diagnosis.
-
Conclusions
- To the best of our knowledge, this is the first study to estimate the prevalence and incidence of HD in Korea and the largest HD series in the Asian population. Our analyses might be useful for further studies and large-scale investigations in HD patients.
- Data collection from the Korean Rare Diseases Registry and the National Health Insurance databases
- We collected the data on HD patients who registered in the RDR between July 2009 and December 2013. During this period, the total number of registered HD cases and the number of new registrations each year were identified. To estimate the prevalence of HD, the size of the Korean population in December 2013 was ascertained from resident registration data with respect to population data gathered by the Korean Ministry of Security and Public. The annual incidence of HD from 2010 to 2013 was calculated based on the number of newly registered HD patients and the number of registered residents in December of the corresponding year. The Korean NHI database was used to crosscheck the prevalence of HD, based on the number of patients receiving medical care.
- Data collection from 10 referral hospitals around the country
- For analysis of the clinical data of individual HD patients, medical records of 68 HD patients from 10 referral hospitals around the country were reviewed. All patients had been diagnosed by a genetic test, which confirmed pathologically expanded CAG repeats at the HTT locus. There were no duplicate cases. The collected clinical data included patient demographics, the number of expanded and normal CAG repeats, initial symptoms and age of onset, the cause of the hospital visit (i.e., the chief complaints), the date of the hospital visit for neurological examination, and the age at genetic diagnosis. There were sometimes multiple initial symptoms and chief complaints for a single patient. Depending on the age of initial symptoms, patients were categorized into juvenile-onset (i.e., HD before the age of 21 years), adult-onset (21–59) and elderly (after 60 years) according to the definition of juvenile-onset HD and the characteristic onset age of typical HD [8,9]. This research was approved by the Institutional Review Board of CHA University.
- Statistical analysis
- The relationship between CAG repeat length and age at initial symptom onset was determined by nonlinear regression analysis. The differences associated with family history and the affected parental sex were compared by independent t test. All of the statistical analyses were performed using Predictive Analytics Software (version 21.0, IBM, New York, NY, USA).
MATERIALS & METHODS
- Crude prevalence and incidence of HD based on RDR and National Health Insurance databases (Table 1)
- Excluding the HD patients registered in 2009 when the RDR program began, the mean number of new patients registered annually between 2010 and 2013 was 27.25 ± 7.14. Based on the resident registration data, the mean of calculated crude annual incidences was 0.06 ± 0.01 cases per 100,000 persons. The estimated crude prevalence of HD was 0.41 cases per 100,000 persons, based on the cumulative number of HD cases in the RDR database (n = 208) and the size of the Korean population (n = 51,141,463) as of December 2013. According to the NHI database, the mean number of HD patients receiving medical services annually between 2009 and 2013 was 190.60 ± 19.86 (ranges from 171 to 222), with the mean percentage of males being 43.42 ± 4.48%. The crude prevalence estimated based on the NHI database was 0.38 ± 0.04.
- Analyses of HD patient data collected from 10 referral hospitals around the country
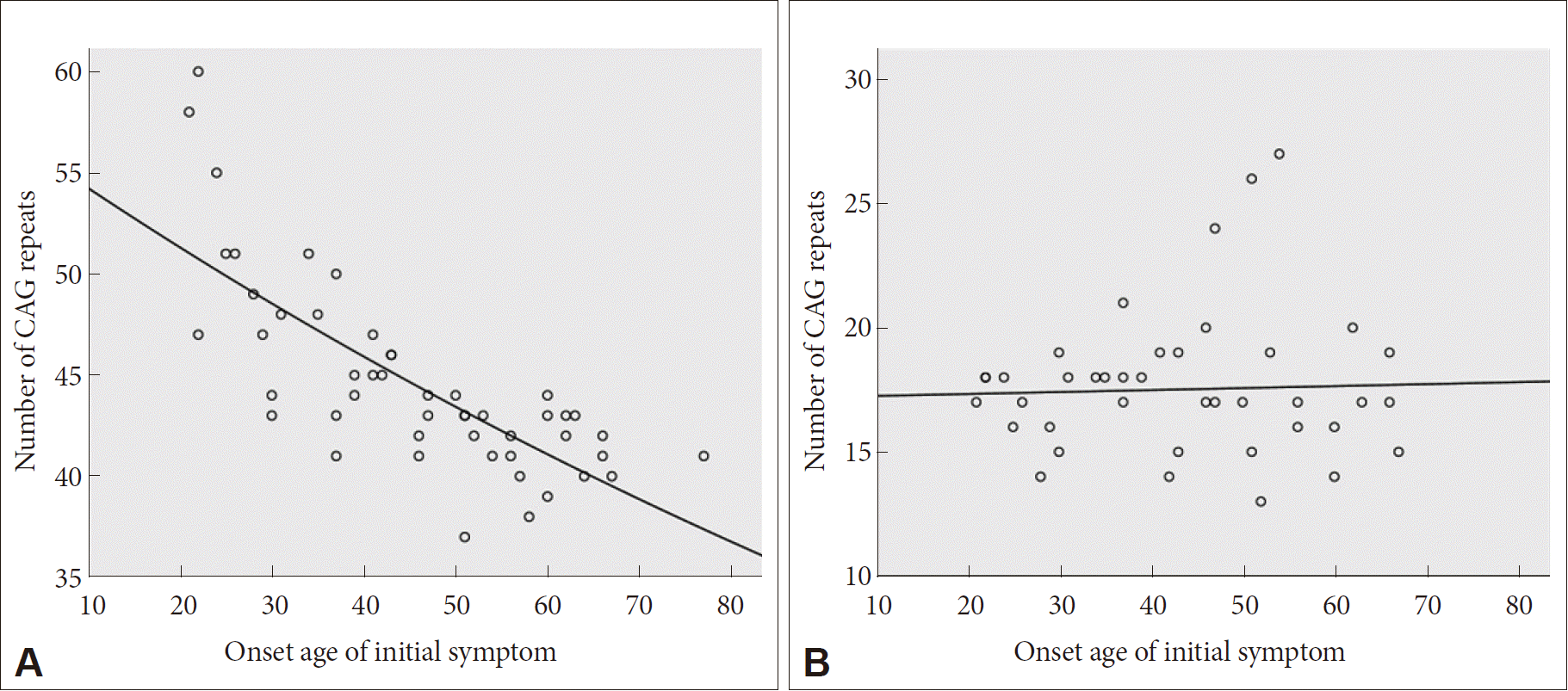
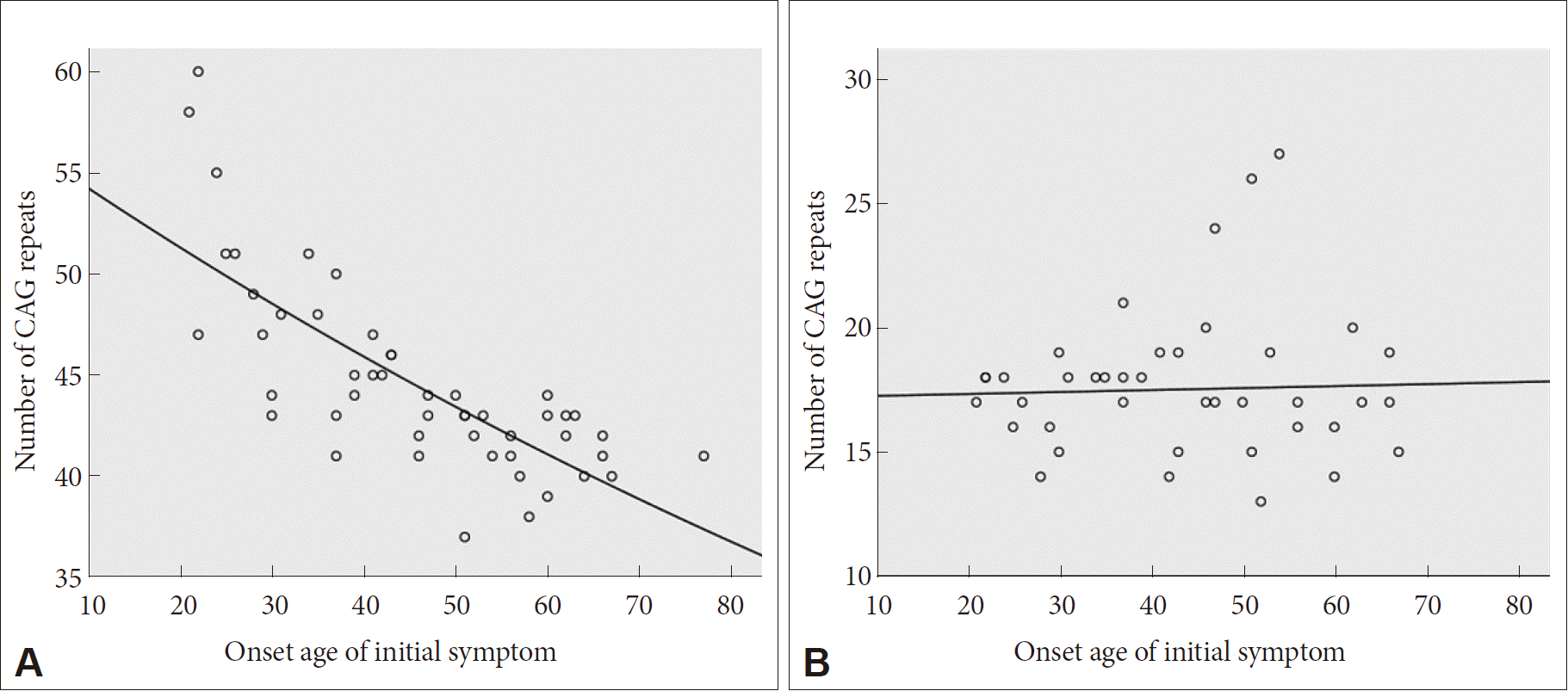
- Sixty-eight HD patients, including 12 patients from six families, were recruited for this study. The mean number of CAG repeats of the expanded allele was 44.7 ± 4.8. In normal alleles, the mean number of CAG repeats was 17.4 ± 3.2 without skewness in the distribution (data not shown). The number of CAG expansion showed an inverse correlation with the onset age of initial symptoms (R2 = 0.574, p-value < 0.001), and there was no correlation between the onset age of initial symptoms and the number of CAG repeats in normal allele (Figure 1). The mean age at initial symptom onset was 44.16 ± 14.08 years old, and the mean interval from initial symptom onset to the diagnosis of HD was 4.30 ± 2.96 years. Chorea (n = 41, 60.3%) was the most frequent initial symptom followed by psychiatric symptoms (n = 19), gait disturbance (n = 13), dysarthria (n = 9), cognitive impairment (n = 8), parkinsonism (n = 2), and urinary symptom (n = 1). Causes of hospital visit (chief complaints) were chorea (n = 58, 85.3%), gait disturbance (n = 18), psychiatric symptoms (n = 16), cognitive impairment (n = 12), dysarthria (n = 10), parkinsonism (n = 4), tremor (n = 3), limb ataxia (n = 2), dysphagia (n = 1), and dystonia (n = 1). The age at initial symptom onset was identified in 62 patients, among whom there was 1 juvenile-onset patient (1.6%) and 8 elderly-onset patients (12.8%). The occupations of 50 patients (73.5%) were classified based on the Korean standard classification of occupations with the addition of several categories such as “no employment” and “housewife”. There was no significant correlation between HD diagnosis and occupation (Figure 2).
- Positive family history of HD was found in 43 patients (63.2%), with paternal transmission occurring in 26 patients (60.5%) and maternal transmission in 17 patients (39.5%). In 21 patients (30.9%), there was no identifiable family history. Four patients had possible family histories, having had family members with early or unexpected mortality associated with psychiatric symptoms and abnormal movements. The comparison of clinical variables between HD patients with and without family histories showed a delay in the onset of initial symptoms (p = 0.047), a prolonged interval between initial symptom onset and genetic diagnosis (p = 0.031) and a delay of the age at genetic diagnosis (p = 0.025) in those without a positive family history (Table 3). Neither the number of expanded CAG repeats nor the interval between the onset of chief complaints and hospital visit was different between the HD patients with and without family histories.
RESULTS
Genetic analysis and clinical features (Table 2)
Comparisons according to the family history (Table 3)
- To the best of our knowledge, this is the first study to estimate the prevalence and incidence of HD in Korea and the largest HD series of multi-center study in the Asian population. Most of the previous Asian epidemiologic studies of HD were performed on a regional basis, such as in Hong Kong, China or the San-in area of Japan [10,11]. In Taiwan, the average annual incidence rate was estimated at 0.1 and the prevalence at 0.42 per 100,000 persons, based on the National Health Insurance database [12]. The annual incidence estimated in our study is about half of the average annual incidence of Taiwan; however, our estimate is similar to the annual incidence in China (0.046 per 100,000) [10]. Our estimation of the prevalence of HD in Korea based on two data sources was 0.41 per 100,000 in 2013 (Korean RDR) and 0.38 per 100,000 bewteen 2009 to 2013 (National Health Insurance database), which are comparable to Taiwanese data and a meta-analysis [11,13]. It is known that HD is less prevalent in Asian countries compared with Western countries, in which the overall prevalence is 5.70 according to a meta-analysis [13]. Recent data suggests that the prevalence of HD may be over 10 per 100,000 in the UK [14]. These racial differences in prevalence are accounted for by the differences in CAG tract size, HTT haplotype and CCG polymorphism [15–17]. The mean CAG repeat length of the normal allele was reported to be significantly longer in the European (18.4 ± 3.7) population compared with the Chinese, Japanese, and Thai (16.4 ± 1.5, 16.6 ± 1.5, and 16.5 ± 1.9, respectively) population [16,18]. Furthermore, the distribution of CAG length was positively skewed in Europeans, which represents a length-dependent mutational bias towards longer alleles [19]. In our collected dataset, we did not observe skewness in CAG length of normal alleles. The mean CAG repeat length of the normal allele in our HD patients was 17.4 ± 3.2, which is longer than that of other Asian populations, but shorter than that reported by a previous single center study in Korea [20] and than those of Western populations. Recently, HTT haplogroups (types A, B, and C) were identified, and CAG instability was more likely to occur in the cis-acting haplogroup A [15]. HTT haplogroups are known to show a regional difference, and the higher risk haplotype was infrequent in China, Japan, and Thailand [18,21,22]. The CCG polymorphic region is located adjacent to the CAG triplet repeat of the HD gene, and there is a linkage disequilibrium between CCG polymorphism and the pathological CAG expansion according to the race [17,23]. HTT haplogroup and CCG polymorphism have not yet been studied in Korean HD patients.
- The mean age at initial symptom onset (44.16 ± 14.08) and the inverse correlation between age at initial symptom onset and CAG repeat expansion size were compatible with previous reports [20,24–26]. The mean length of CAG repeats were similar to the previously reported range of 44.7 to 49.0 (range: 36–70) in Korea and Western countries [5–7,20,27]. Moreover, the length of the expanded CAG repeat determines the age of onset in approximately 50–70% of patients in the 40 to 55 CAG repeat range, but possible roles of other genetic and environmental modifiers are currently being investigated [28,29].
- Chorea is the most frequent initial symptom (60.3%) in our collected dataset as well as the chief complaint (85.3%), and the latter is compatible to the previously reported frequency (89%) at first visit to hospital [20]. However, this finding also showed that approximately 40% of HD patients had initial symptoms other than chorea. Among the symptomatic triad of problems involving movement, cognition, and mood, the most prominent deficit may differ between patients, and recent studies have recognized that non-motor symptoms occur before overt motor features in a fraction of patients [30,31].
- About one third of the patients comprising our collected dataset had no family history of HD, which is higher than reported in a study from British Columbia (29.8%) [32] and in a previous report from a single center in Korea (20%).20 Given that the probability of de novo expansion of CAG repeats through paternal transmission was estimated to range from 1/6,241 to 1/951 [33], the higher rate of HD patients with a negative family history might not be explained solely by this mechanism. In addition to de novo expansion of CAG repeats, unstable familial transmission, reduced penetrance or underreported family history may be another cause of negative family history among HD patients [34–36]. Interestingly, our HD patients without family history showed a delayed age at onset of initial symptoms, a delayed age at genetic diagnosis and a prolonged interval between the age of initial symptom onset to the age of genetic diagnosis compared with the HD patients with a positive family history. Lower CAG repeat number in negative family history groups may be related with these findings; however, a clinician’s delay in suspicion of HD in choreic patients apparently lacking family history may have also contributed to this observation. Our study has some limitations. Because this research is based on retrospective chart review, extensive data gathering was impossible. Even after the diagnosis of HD, some patients may have refused to be registered into RDR because of social stigma and discrimination, which may have resulted in the underestimation of the real incidence and prevalence of HD [37].
- In conclusion, our estimated incidence and prevalence of HD were comparable with previous reports of Asian populations and were relatively low compared with estimates for Western populations. The clinical features showed similar phenotypes, and the effect of CAG repeat expansion on the age of disease onset was comparable. Our analyses using national registries and multi-center clinical data could be useful for future studies and large-scale investigations in HD patients.
DISCUSSION

Total Korean population taken from resident registration data (December of each year). Estimated annual incidence = (incident cases per year / total Korean population) × 105, Estimated prevalence = (total cases per year / total Korean population) × 105. HD: Huntington’s disease, NA: not available, NHI: National Health Insurance, RDR: Rare Diseases Registry.
| Clinical findings | Family history | p-value | |
|---|---|---|---|
| Presence | Absence | ||
| Age at initial symptom onset | 41.81 ± 13.40 | 49.47 ± 14.49 | 0.047* |
| Interval between the age at initial symptom onset to the genetic diagnosis (years) | 3.80 ± 2.71 | 5.58 ± 3.21 | 0.031* |
| Age at genetic diagnosis (years) | 45.40 ± 13.64 | 54.11 ± 14.79 | 0.025* |
| Interval between the onset of chief complaint to hospital visit (months) | 29.45 ± 23.76 | 43.59 ± 38.08 | 0.17 |
| Number of expanded CAG repeat | 45.30 ± 5.14 | 43.41 ± 3.64 | 0.18 |
| Number of normal CAG repeat | 17.28 ± 3.49 | 17.83 ± 2.48 | 0.62 |
- 1. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993;72:971–983.ArticlePubMed
- 2. Lyoo CH, Lee MS, Kim YJ, Suk SH, Yang KH, Song KS. Huntington’s disease confirmed by genetic and pathological study. J Korean Neurol Assoc 1996;14:725–737.
- 3. Kim BJ, Park KW, Lee DH. Diagnosis of Huntington’s disease with polymerase chain reaction. J Korean Neurol Assoc 1996;14:502–510.
- 4. Jeon BS, Choi SH, Kim MH, Joo SI, Park SS. Gene analysis in Huntington disease. J Korean Neurol Assoc 1996;14:494–501.
- 5. Kim SY, Park SS, Joo SI, Lee DH, Wie BA, Kwon HY, et al. Clinical analysis of Huntington’s disease in Korea. J Korean Neurol Assoc 1997;15:1256–1264.
- 6. Shin H, Kim MH, Lee SJ, Lee KH, Kim MJ, Kim JS, et al. Decreased metabolism in the cerebral cortex in early-stage Huntington’s disease: a possible biomarker of disease progression? J Clin Neurol 2013;9:21–25.ArticlePubMedPMC
- 7. Bates G, Tabrizi S, Jones L. Huntington’s Disease. 4th ed. New York: Oxford University Press; 2014.
- 8. Hille ET, Siesling S, Vegter-van der Vlis M, Vandenbroucke JP, Roos RA, Rosendaal FR. Two centuries of mortality in ten large families with Huntington disease: a rising impact of gene carriership. Epidemiology 1999;10:706–710.ArticlePubMed
- 9. Telenius H, Kremer HP, Theilmann J, Andrew SE, Almqvist E, Anvret M, et al. Molecular analysis of juvenile Huntington disease: the major influence on (CAG)n repeat length is the sex of the affected parent. Hum Mol Genet 1993;2:1535–1540.ArticlePubMedPDF
- 10. Chang CM, Yu YL, Fong KY, Wong MT, Chan YW, Ng TH, et al. Huntington’s disease in Hong Kong Chinese: epidemiology and clinical picture. Clin Exp Neurol 1994;31:43–51.PubMed
- 11. Nakashima K, Watanabe Y, Kusumi M, Nanba E, Maeoka Y, Nakagawa M, et al. Epidemiological and genetic studies of Huntington’s disease in the San-in area of Japan. Neuroepidemiology 1996;15:126–131.ArticlePubMed
- 12. Chen YY, Lai CH. Nationwide population-based epidemiologic study of Huntington’s Disease in Taiwan. Neuroepidemiology 2010;35:250–254.ArticlePubMed
- 13. Pringsheim T, Wiltshire K, Day L, Dykeman J, Steeves T, Jette N. The incidence and prevalence of Huntington’s disease: a systematic review and meta-analysis. Mov Disord 2012;27:1083–1091.ArticlePubMed
- 14. Evans SJ, Douglas I, Rawlins MD, Wexler NS, Tabrizi SJ, Smeeth L. Prevalence of adult Huntington’s disease in the UK based on diagnoses recorded in general practice records. J Neurol Neurosurg Psychiatry 2013;84:1156–1160.ArticlePubMedPMC
- 15. Warby SC, Visscher H, Collins JA, Doty CN, Carter C, Butland SL, et al. HTT haplotypes contribute to differences in Huntington disease prevalence between Europe and East Asia. Eur J Hum Genet 2011;19:561–566.ArticlePubMedPMC
- 16. Squitieri F, Andrew SE, Goldberg YP, Kremer B, Spence N, Zeisler J, et al. DNA haplotype analysis of Huntington disease reveals clues to the origins and mechanisms of CAG expansion and reasons for geographic variations of prevalence. Hum Mol Genet 1994;3:2103–2114.ArticlePubMedPDF
- 17. Pêcheux C, Mouret JF, Dürr A, Agid Y, Feingold J, Brice A, et al. Sequence analysis of the CCG polymorphic region adjacent to the CAG triplet repeat of the HD gene in normal and HD chromosomes. J Med Genet 1995;32:399–400.ArticlePubMedPMC
- 18. Pulkes T, Papsing C, Wattanapokayakit S, Mahasirimongkol S. CAG-Expansion Haplotype Analysis in a Population with a Low Prevalence of Huntington’s Disease. J Clin Neurol 2014;10:32–36.ArticlePubMedPMC
- 19. Rubinsztein DC, Amos W, Leggo J, Goodburn S, Ramesar RS, Old J, et al. Mutational bias provides a model for the evolution of Huntington’s disease and predicts a general increase in disease prevalence. Nat Genet 1994;7:525–530.ArticlePubMedPDF
- 20. Shin CW, Choi YJ, Kim M, Jeon BS. Preliminary analysis of Huntington’s Disease in South Korea. J Huntingtons Dis 2013;2:83–87.ArticlePubMed
- 21. Masuda N, Goto J, Murayama N, Watanabe M, Kondo I, Kanazawa I. Analysis of triplet repeats in the huntingtin gene in Japanese families affected with Huntington’s disease. J Med Genet 1995;32:701–705.ArticlePubMedPMC
- 22. Ma M, Yang Y, Shang H, Su D, Zhang H, Ma Y, et al. Evidence for a predisposing background for CAG expansion leading to HTT mutation in a Chinese population. J Neurol Sci 2010;298:57–60.ArticlePubMed
- 23. Morovvati S, Nakagawa M, Osame M, Karami A. Analysis of CCG repeats in Huntingtin gene among HD patients and normal populations in Japan. Arch Med Res 2008;39:131–133.ArticlePubMed
- 24. Aziz NA, Jurgens CK, Landwehrmeyer GB. EHDN Registry Study Group. van Roon-Mom WM, van Ommen GJ, et al. Normal and mutant HTT interact to affect clinical severity and progression in Huntington disease. Neurology 2009;73:1280–1285.ArticlePubMed
- 25. Brinkman RR, Mezei MM, Theilmann J, Almqvist E, Hayden MR. The likelihood of being affected with Huntington disease by a particular age, for a specific CAG size. Am J Hum Genet 1997;60:1202–1210.PubMedPMC
- 26. Langbehn DR, Brinkman RR, Falush D, Paulsen JS, Hayden MR. International Huntington’s Disease Collaborative Group. A new model for prediction of the age of onset and penetrance for Huntington’s disease based on CAG length. Clin Genet 2004;65:267–277.ArticlePubMed
- 27. Orth M, Schwenke C. Age-at-onset in Huntington disease. PLoS Curr 2011;3:RRN1258.ArticlePubMedPMC
- 28. Gusella JF, MacDonald ME, Lee JM. Genetic modifiers of Huntington’s disease. Mov Disord 2014;29:1359–1365.ArticlePubMed
- 29. Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 2011;10:83–98.ArticlePubMed
- 30. Paulsen JS, Long JD. Onset of Huntington’s disease: can it be purely cognitive? Mov Disord 2014;29:1342–1350.ArticlePubMedPMC
- 31. Tabrizi SJ, Scahill RI, Durr A, Roos RA, Leavitt BR, Jones R, et al. Biological and clinical changes in premanifest and early stage Huntington’s disease in the TRACK-HD study: the 12-month longitudinal analysis. Lancet Neurol 2011;10:31–42.ArticlePubMed
- 32. Almqvist EW, Elterman DS, MacLeod PM, Hayden MR. High incidence rate and absent family histories in one quarter of patients newly diagnosed with Huntington disease in British Columbia. Clin Genet 2001;60:198–205.ArticlePubMed
- 33. Hendricks AE, Latourelle JC, Lunetta KL, Cupples LA, Wheeler V, MacDonald ME, et al. Estimating the probability of de novo HD cases from transmissions of expanded penetrant CAG alleles in the Huntington disease gene from male carriers of high normal alleles (27–35 CAG). Am J Med Genet A 2009;149A:1375–1381.ArticlePubMedPMC
- 34. Semaka A, Collins JA, Hayden MR. Unstable familial transmissions of Huntington disease alleles with 27–35 CAG repeats (intermediate alleles). Am J Med Genet B Neuropsychiatr Genet 2010;153B:314–320.Article
- 35. Houge G, Bruland O, Bjørnevoll I, Hayden MR, Semaka A. De novo Huntington disease caused by 26–44 CAG repeat expansion on a low-risk haplotype. Neurology 2013;81:1099–1100.ArticlePubMedPMC
- 36. Sequeiros J, Ramos EM, Cerqueira J, Costa MC, Sousa A, Pinto-Basto J, et al. Large normal and reduced penetrance alleles in Huntington disease: instability in families and frequency at the laboratory, at the clinic and in the population. Clin Genet 2010;78:381–387.ArticlePubMed
- 37. Dispelling the stigma of Huntington’s disease. Lancet Neurol 2010;9:751.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
- Analysis of HTT CAG repeat expansion among healthy individuals and patients with chorea in Korea
Ryul Kim, Moon-Woo Seong, Bumjo Oh, Ho Seop Shin, Jee-Soo Lee, Sangmin Park, Mihee Jang, Beomseok Jeon, Han-Joon Kim, Jee-Young Lee
Parkinsonism & Related Disorders.2024; 118: 105930. CrossRef - Epidemiology of Chronic Inflammatory Demyelinating Polyneuropathy in South Korea: A Population-Based Study
Sohee Jung, Gucheol Jung, Dayoung Kim, Jeeyoung Oh, Kyomin Choi
Journal of Clinical Neurology.2023; 19(6): 558. CrossRef - Increased 10-Year Prevalence of Huntington’s Disease in South Korea: An Analysis of Medical Expenditure Through the National Healthcare System
Chan Young Lee, Jun-soo Ro, Hyemin Jung, Manho Kim, Beomseok Jeon, Jee-Young Lee
Journal of Clinical Neurology.2023; 19(2): 147. CrossRef - Global Epidemiology of Movement Disorders: Rare or Underdiagnosed?
Sarah A. O'Shea, Ludy C. Shih
Seminars in Neurology.2023; 43(01): 004. CrossRef - Huntington’s Disease in Chile: Epidemiological and Genetic Aspects
Ernesto Solís-Añez, Philippe A. Salles, Natalia Rojas, Olga Benavides, Pedro Chaná-Cuevas
Neuroepidemiology.2023; 57(3): 176. CrossRef - Epidemiology of Acute Leukemia among Children with Down Syndrome in Korea
Young Bae Choi, Keon Hee Yoo
Cancer Research and Treatment.2022; 54(2): 572. CrossRef - Population Prevalence, Cancer Risk, and Mortality Risk of Turner Syndrome in South Korean Women Based on National Health Insurance Service Data
Sung Eun Kim, Sang Hyun Park, Kyungdo Han, Won Kyoung Cho, Byung-Kyu Suh, Yong-Gyu Park
Yonsei Medical Journal.2022; 63(11): 991. CrossRef - Prevalence and Incidence of Huntington's Disease: An Updated Systematic Review and Meta‐Analysis
Alex Medina, Yasamin Mahjoub, Larry Shaver, Tamara Pringsheim
Movement Disorders.2022; 37(12): 2327. CrossRef - Contemporary Status of Acute Myocardial Infarction in Korean Patients: Korean Registry of Acute Myocardial Infarction for Regional Cardiocerebrovascular Centers
Rock Bum Kim, Jin Yong Hwang, Hyun Woong Park, Ae-Young Her, Jang Hoon Lee, Moo Hyun Kim, Chang Hwan Yoon, Jae Young Cho, Sung-Il Woo, Yongcheol Kim, Jae-Young Han, Joon Hyouk Choi, Song Yi Kim, Si Wan Choi, Sung Ju Jee, Sang Yeub Lee, Ki-Bum Won, Kyeong-
Journal of Clinical Medicine.2021; 10(3): 498. CrossRef - Huntington’s Disease in Israel: A Population-Based Study Using 20 Years of Routinely-Collected Healthcare Data
Natalie Gavrielov-Yusim, Yael Barer, Michael Martinec, Athanasios Siadimas, Spyros Roumpanis, Hannah Furby, Inbal Goldshtein, Asif Jan, Preciosa M. Coloma
Journal of Huntington's Disease.2021; 10(4): 469. CrossRef The Population Prevalence, Associations of Congenital Heart Defect and Mortality Risk for Down’s Syndrome in South Korea Based on National Health Insurance Service (NHIS) Data
Won Kyoung Cho, Na Young Lee, Kyungdo Han, Byung-Kyu Suh, Yong-Gyu Park
Clinical Epidemiology.2020; Volume 12: 519. CrossRef- Incidence of Huntington disease in a northeastern Spanish region: a 13-year retrospective study at tertiary care centre
Paula Sienes Bailo, Raquel Lahoz, Juan Pelegrín Sánchez Marín, Silvia Izquierdo Álvarez
BMC Medical Genetics.2020;[Epub] CrossRef - The molecular epidemiology of Huntington disease is related to intermediate allele frequency and haplotype in the general population
Chris Kay, Jennifer A. Collins, Galen E.B. Wright, Fiona Baine, Zosia Miedzybrodzka, Folefac Aminkeng, Alicia J. Semaka, Cassandra McDonald, Mark Davidson, Steven J. Madore, Erynn S. Gordon, Norman P. Gerry, Mario Cornejo‐Olivas, Ferdinando Squitieri, Sar
American Journal of Medical Genetics Part B: Neuropsychiatric Genetics.2018; 177(3): 346. CrossRef - Quantitative Gait Analysis in Patients with Huntington’s Disease
Seon Jong Pyo, Hanjun Kim, Il Soo Kim, Young-Min Park, Mi-Jung Kim, Hye Mi Lee, Seong-Beom Koh
Journal of Movement Disorders.2017; 10(3): 140. CrossRef - Epidemiological Study of Huntington's Disease in the Province of Ferrara, Italy
Erika Carrassi, Maura Pugliatti, Vittorio Govoni, Mariachiara Sensi, Ilaria Casetta, Enrico Granieri
Neuroepidemiology.2017; 49(1-2): 18. CrossRef - The Epidemiology of Myasthenia Gravis in Korea
Hyung Seok Lee, Hye Sun Lee, Ha Young Shin, Young-Chul Choi, Seung Min Kim
Yonsei Medical Journal.2016; 57(2): 419. CrossRef - The global prevalence of Huntington's disease: a systematic review and discussion
Sheharyar Sajjad Baig, Mark Strong, Oliver WJ Quarrell
Neurodegenerative Disease Management.2016; 6(4): 331. CrossRef - Clinical and genetic characteristics in patients with Huntington’s disease from China
Jing Yang, Ke Chen, Qianqian Wei, Yongping Chen, Bei Cao, Jean-Marc Burgunder, Hui-Fang Shang
Neurological Research.2016; 38(10): 916. CrossRef - Survival of Korean Huntington’s Disease Patients
Han-Joon Kim, Chae-Won Shin, Beomseok Jeon, Hyeyoung Park
Journal of Movement Disorders.2016; 9(3): 166. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite