E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 11(1); 2018 > Article
-
Review Article
Tau Positron Emission Tomography Imaging in Degenerative Parkinsonisms - Chul Hyoung Lyoo1, Hanna Cho1, Jae Yong Choi2,3, Young Hoon Ryu2, Myung Sik Lee1
-
Journal of Movement Disorders 2018;11(1):1-12.
DOI: https://doi.org/10.14802/jmd.17071
Published online: January 23, 2018
1Department of Neurology, Gangnam Severance Hospital, Yonsei University College of Medicine, Seoul, Korea
2Department of Nuclear Medicine, Gangnam Severance Hospital, Yonsei University College of Medicine, Seoul, Korea
3Division of RI-Convergence Research, Korea Institute Radiological and Medical Sciences, Seoul, Korea
- Corresponding author: Chul Hyoung Lyoo, MD, PhD, Department of Neurology, Gangnam Severance Hospital, Yonsei University College of Medicine, Research Center for Future Medicine, 20 Eonju-ro 63-gil, Gangnam-gu, Seoul 06229, Korea Tel: +82-2-2019-3326 Fax: +82-2-3462-5904 E-mail: lyoochel@yuhs.ac
• Received: October 16, 2017 • Accepted: November 20, 2017
Copyright © 2018 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- In recent years, several radiotracers that selectively bind to pathological tau proteins have been developed. Evidence is emerging that binding patterns of in vivo tau positron emission tomography (PET) studies in Alzheimer’s disease (AD) patients closely resemble the distribution patterns of known neurofibrillary tangle pathology, with the extent of tracer binding reflecting the clinical and pathological progression of AD. In Lewy body diseases (LBD), tau PET imaging has clearly revealed cortical tau burden with a distribution pattern distinct from AD and increased cortical binding within the LBD spectrum. In progressive supranuclear palsy, the globus pallidus and midbrain have shown increased binding most prominently. Tau PET patterns in patients with corticobasal syndrome are characterized by asymmetrical uptake in the motor cortex and underlying white matter, as well as in the basal ganglia. Even in the patients with multiple system atrophy, which is basically a synucleinopathy, 18F-flortaucipir, a widely used tau PET tracer, also binds to the atrophic posterior putamen, possibly due to off-target binding. These distinct patterns of tau-selective radiotracer binding in the various degenerative parkinsonisms suggest its utility as a potential imaging biomarker for the differential diagnosis of parkinsonisms.
- 11C-Pittsburgh compound B (11C-PIB) is a radiotracer that selectively binds to amyloid-β (Aβ) in senile plaques, which are a pathological hallmark of Alzheimer’s disease (AD). This radiotracer has enabled a new era of pathology-targeted molecular imaging of neurodegenerative diseases. The recent development of 18F-labelled radiotracers that are selective for Aβ, including 18F-flutemetamol, 18F-florbetapir, 18F-florbetaben, and 18F-NAV4694 (formerly 18F-AZD4694), has also facilitated the application of Aβ-imaging for clinical use [1-4]. Positron emission tomography (PET) using these Aβ-selective radiotracers clearly mirrors the extent of Aβ accumulation in the brain [5,6], thereby enabling an earlier diagnosis of prodromal AD [7,8]. However, because neocortical Aβ pathology generally plateaus at an early stage of AD [9], Aβ-imaging is less effective in delineating the progression of AD [10].
- Paired helical filaments (PHF) of hyperphosphorylated tau protein are a major constituent of neurofibrillary tangles (NFT), the second major pathological hallmark of AD [11]. NFTs first appear in the transentorhinal region, spreading hierarchically to the neighboring limbic areas and distant association neocortices before finally reaching the primary cortices [11]. Because the distant propagation of tau pathology is preceded by an early and widespread dissemination of Aβ pathology in the neocortex [9,12], cortical tau burden is a better indicator of the clinical progression of AD [13,14]. In addition, in contrast to the limited number of Aβ-related diseases, the existence of a wider clinical spectrum of tauopathies has necessitated the development of molecular imaging biomarkers for tau protein [15].
- The development of the first tau-selective radiotracer, 18F-THK523, in 2011 was another major breakthrough [16]. Although this PET radiotracer is currently no longer used for clinical research due to serious drawbacks that occurred in human studies [17], it encouraged the development of better tau PET radiotracers that are now used in clinical research (Figure 1).
- Over recent years, clinical tau PET studies have primarily focused on the AD spectrum. Tau PET allows clear visualization of AD tau pathology with a high selectivity for PHF-tau [18,19] and is now generally accepted as a useful imaging biomarker for assessing the pathological and clinical progression of AD [20-22]. In contrast to AD, postmortem autoradiography and a smaller number of in vivo tau PET studies in non-AD tauopathies have consistently reported weaker radiotracer binding to non-AD tau than to PHF-tau in AD [18,19,23-32].
- In this review, we focus on recent progress in the knowledge of tau-selective tracers and clinical tau PET studies in degenerative parkinsonisms, such as Parkinson’s disease (PD), dementia with Lewy bodies (DLB), progressive supranuclear palsy (PSP), corticobasal syndrome (CBS), and multiple system atrophy (MSA).
INTRODUCTION
- 18F-THK series (18F-THK-523, 18F-THK-5105, 18F-THK-5317, and 18F-THK-5351)
- The first tau-selective radiotracer, 18F-THK523, exhibited a 10-fold stronger binding affinity to pathological tau protein than to Aβ fibrils in vitro, selective binding to PHF-tau pathology in autoradiography studies with postmortem AD tissue, and stronger uptake in the brains of tau transgenic mice when compared to the wild-type or APP/PS1 transgenic mice [16]. In contrast to these promising results, subsequent in vivo human PET studies with 18F-THK523 were quite disappointing due to high levels of white matter binding and low standardized uptake value ratio (SUVR) values, even in AD patients [17]. Regional differences in 18F-THK523 binding in AD were only discernible with partial volume correction of the PET images. This regional difference was almost eliminated without the correction. Therefore, 18F-THK523 has been deemed unsuitable for clinical tau PET imaging studies [17]. To overcome these issues, improvements to the 18F-THK series have focused on reducing white matter binding, and the second generation of the 18F-THK series, namely, 18F-THK-5117, 18F-THK-5317, and 18F-THK-5351, have exhibited much lower white matter binding than their predecessor. The most recently developed radiotracer in the 18F-THK series is 18F-THK-5351, which has a higher affinity for PHF-tau and more rapid washout from white matter than the previous version, 18F-THK-5117 [33]. For this reason, 18F-THK-5351 PET achieves higher contrasts between true binding and background and a much lower degree of white matter binding than 18F-THK-5117 and is now considered a useful imaging biomarker for AD.
- Even with these positive findings for 18F-THK-5351, white matter binding is still a significant issue in comparison to other tau-selective radiotracers [17]. High white matter binding may mask small increases in 18F-THK-5351 binding in the gray matter due to an overflow of PET signals from the adjacent white matter. Likewise, 18F-THK-5351 PET still exhibits elevated binding in the pons. This may affect accurate determination of tau pathology in the brainstem. Additionally, similar to 18F-flortaucipir, off-target binding to the basal ganglia, even in healthy elderly individuals, is another common issue for the 18F-THK series.
- A recent 18F-THK-5351 PET study reported serious problems relating to monoamine oxidase-B (MAO-B) binding [34]. MAO-B is widely expressed in the brain, most prominently in the basal ganglia, followed by the insular cortex [35]. This topographical pattern was replicated in several in vivo PET studies [36-38]. In healthy controls and patients with mild cognitive impairment, AD, and PSP, one study conducted three 18F-THK-5351 PET scans acquired before and after 10 mg of the MAO-B inhibitor, selegiline, was orally administered and again at 9–28 days after the selegiline treatment [34]. Surprisingly, a single oral dose of selegiline dramatically reduced 18F-THK-5351 standardized uptake values by 37–52% across all regions, most prominently in the thalamus (52%) and basal ganglia (51%), and even in the cerebellar cortex (42%), which is generally used as a reference tissue. This suppressive effect was sustained until the third PET scan [34]. Therefore, the MAO-B binding characteristics of 18F-THK-5351 may limit its applicability in tau imaging.
- 18F-flortaucipir (formerly referred to as 18F-AV-1451 or 18F-T807)
- 18F-flortaucipir has exhibited a 25-fold greater binding affinity to PHF-tau than to Aβ, and very low white matter binding in several in vivo human PET studies [39]. As a result, 18F-flortaucipir PET enables high contrasts between binding and background, which are helpful for detecting small increases in cortical binding. Unlike the similar radiotracer, 18F-T808, which shows a high skull uptake in some subjects due to serious defluorination [40], 18F-flortaucipir does not exhibit defluorination issues in human [39,41]. Due to these positive findings, 18F-flortaucipir has been most widely used for clinical tau imaging studies.
- Autoradiography studies of postmortem tissues have consistently reported a stronger binding affinity of 18F-flortaucipir to PHF-tau in AD, in contrast to its much weaker binding affinity to straight filament tau in non-AD tauopathies [18,19]. Therefore, 18F-flortaucipir is better for tau imaging studies in AD rather than in various other non-AD tauopathies.
- However, there are two significant issues with 18F-flortaucipir. First, unlike the other types of tau-selective radiotracers, which show stable SUVR values after a certain time point, 18F-flortaucipir has unstable kinetics, causing the SUVR values to steadily increase even after 60 mins [41]. This characteristic can limit quantification attempts, especially in longitudinal studies [42], although data acquired 80–100 mins post-injection can provide reliable SUVR values that correlate with the binding values determined by compartmental modeling [43,44]. A second problem is the widely reported off-target binding. 18F-flortaucipir also exhibits a high affinity for melanin-producing cells, including the substantia nigra, skin epithelium, retinal pigment epithelium, and melanomas. It, therefore, binds strongly to the substantia nigra, in which a high concentration of neuromelanin exists [18,45]. 18F-flortaucipir also strongly binds to the basal ganglia, even in healthy elderly individuals with an absence of tau pathology [18,45]. One study showed a possible interaction with iron due to a correlation between age-related increases in basal ganglial iron content and 18F-flortaucipir binding in the basal ganglia [46]. Nigral and basal ganglial off-target binding is problematic for tau imaging, especially in parkinsonisms. The choroid plexus is another off-target binding site. Although one study found tangle-like structures that were immunoreactive to phosphorylated tau antibody in the epithelial cells in the choroid plexus [47], the exact mechanism of this off-target binding remains unknown. Off-target binding in the choroid plexus also disturbs the precise quantitation of underlying hippocampal binding and can be an obstacle to early detection of hippocampal tau burden.
- 18F-flortaucipir binds to MAO-A with a high affinity [48], but unlike 18F-THK-5351, there have been no reports to date of 18F-flortaucipir binding to MAO-B.
- 18F-MK6240
- 18F-MK6240 is the most recently developed tau-selective radiotracer, and, thus, there is little information about it. One autoradiographical PET study in monkeys reported a 5-fold higher binding potential of 3H-MK6240, no off-target binding, and no MAO-A binding when compared to 3H-flortaucipir [48,49]. In a small number of healthy elderly subjects and AD patients, 18F-MK6240 exhibited fast washout, high binding to the cortical regions vulnerable to AD pathology, and a good correlation with the severity of cognitive impairment in AD [50]. Larger clinical PET studies are needed to better characterize the 18F-MK6240 radiotracer.
- 11C-PBB3
- Unlike 18F-labelled compounds with longer half-lives (109 mins), the shorter (20 mins) half-life of 11C-labelled compound permits two PET scans in the same day. Therefore, 11C-labelled compounds are suitable for research-based PET, while 18F-labelled compounds are suitable for clinical PET scans. 11C-PBB3 is the only 11C-labelled tau-selective radiotracer that has an approximate 50-fold higher affinity for PHF-tau than that for Aβ [51]. An in vivo 11C-PBB3 PET study also exhibited high tracer binding to the cortical regions of AD, similar to other types of tau-selective radiotracers [51]. More importantly, PBB3 also has a higher affinity for 4-repeat (4R) or 3R tau than 18F-flortaucipir and is considered to be a tau PET tracer specific for a broader range of tau [52]. However, 11C-PBB3 is rapidly metabolized in plasma, and radioactive metabolites that enter into the brain can contaminate PET signals. This problem makes 11C-PBB3 unsuitable for quantification [53,54]. In addition, high tracer retention in the venous sinus in all human subjects may contaminate PET signals around the venous sinus [51].
CHARACTERISTICS OF TAUSELECTIVE RADIOTRACERS
- PD with normal cognition (PDNC), PD with mild cognitive impairment (PDMCI), PD with dementia (PDD), and DLB all share common clinical characteristics and neuropathology and are now considered to be part of the Lewy body diseases (LBD) spectrum [55,56]. In addition to the well-known α-synuclein pathologies presenting as Lewy bodies and Lewy neurites, AD-type pathologies containing Aβ and PHF-tau are also found in LBD [57,58]. Although the prevalence of Aβ-positivity seen in the 11C-PIB PET studies of LBD, can be highly variable [59-64], a clearly increasing trend for the overall prevalence of Aβ-positivity within the LBD spectrum (5% in PDMCI, 34% in PDD, and 68% in DLB) has been observed [65]. Therefore, a similar increasing trend of cortical binding in tau PET studies can be expected.
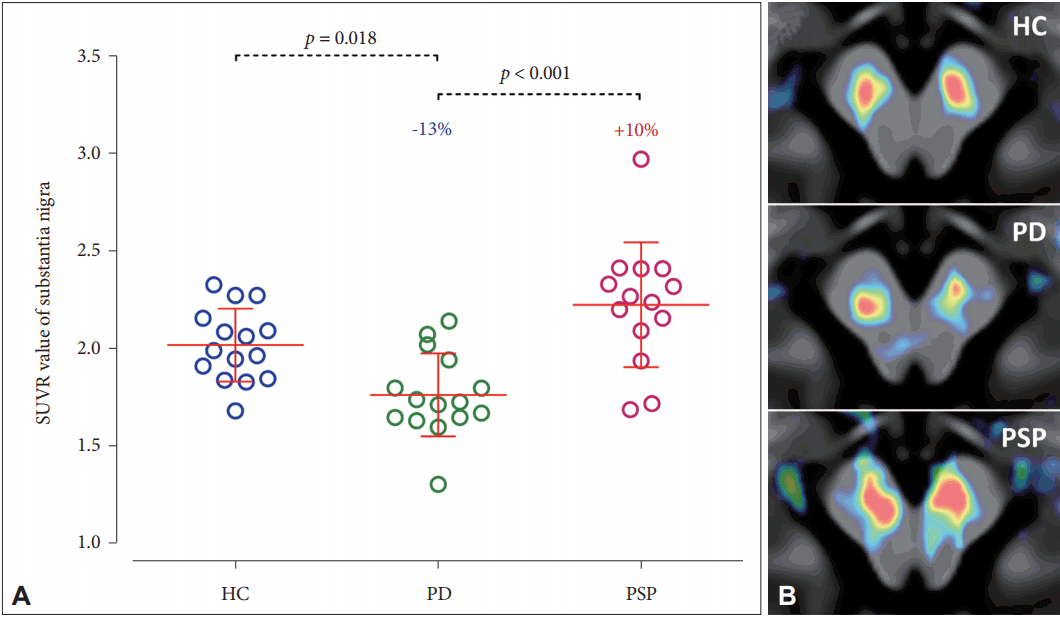
- All 18F-flortaucipir PET studies in PD patients to date have consistently shown no increased binding in the basal ganglia or in the cerebral cortex [25,66-69]. PD patients exhibited approximately 13% lower 18F-flortaucipir binding in the substantia nigra compared to controls [25,66,67], due to off-target binding of 18F-flortaucipir to neuromelanin pigment, which normally exists in the substantia nigra and is lost in PD (Figure 2) [18,19]. Reduced 18F-flortaucipir binding was more prominent in the lateral part of the substantia nigra than in the medial part. However, nigral 18F-flortaucipir binding did not correlate with the motor severity of PD and did not reflect clinical asymmetry [25,66].
- DLB is positioned at the end of the LBD spectrum and can, therefore, be expected to exhibit the greatest cortical 18F-flortaucipir binding. The first 18F-flortaucipir PET study in a small number of patients within the LBD spectrum [7 DLB, 8 PD with cognitive impairment (PDCI), and 9 PDNC] showed an increasing trend of cortical 18F-flortaucipir binding [68]. 18F-flortaucipir binding was increased in the inferior temporal and precuneus cortices in DLB and in the same area in PDCI, with a lower level of statistical significance. The binding in the inferior temporal and precuneus cortices correlated with the severity of cognitive impairment only in the composite group with DLB and PDCI [68].
- The second 18F-flortaucipir PET study involved 19 DLB and 19 AD patients [70]. Compared to the controls, the DLB patients showed greater binding in the posterior temporo-parietal and occipital cortices, in which 18F-flortaucipir binding correlated with the global cortical 11C-PIB binding. Interestingly, the medial temporal regions were relatively preserved in the DLB patients when compared to the AD patients, and for this reason, medial temporal 18F-flortaucipir binding may be useful for differential diagnosis between DLB and AD. However, they found no correlation between the 18F-flortaucipir binding and the severities of cognitive impairment and parkinsonian motor deficits [70].
- A recent 18F-flortaucipir PET study in a larger number of patients within the LBD spectrum (18 DLB, 22 PDCI, and 12 PDNC) showed a clearly increasing trend of cortical 18F-flortaucipir binding within the LBD spectrum [69]. In this report, 18F-flortaucipir binding was dependent on Aβ-positivity, as determined by 18F-florbetaben PET. Compared to the controls, the Aβ-positive DLB group showed significantly increased binding in the sensorimotor, primary visual, and parieto-temporal cortices, and the Aβ-positive PDCI group showed slightly increased binding in the middle and inferior temporal and parahippocampal cortices without surviving multiple comparisons. All Aβ-negative DLB, PDCI, and PDNC groups showed no increased binding in any of the cortical regions. In DLB, there was only a weak correlation between the severity of the cognitive impairment and binding in the prefrontal, sensorimotor, posterior cingulate, and occipital cortices [69].
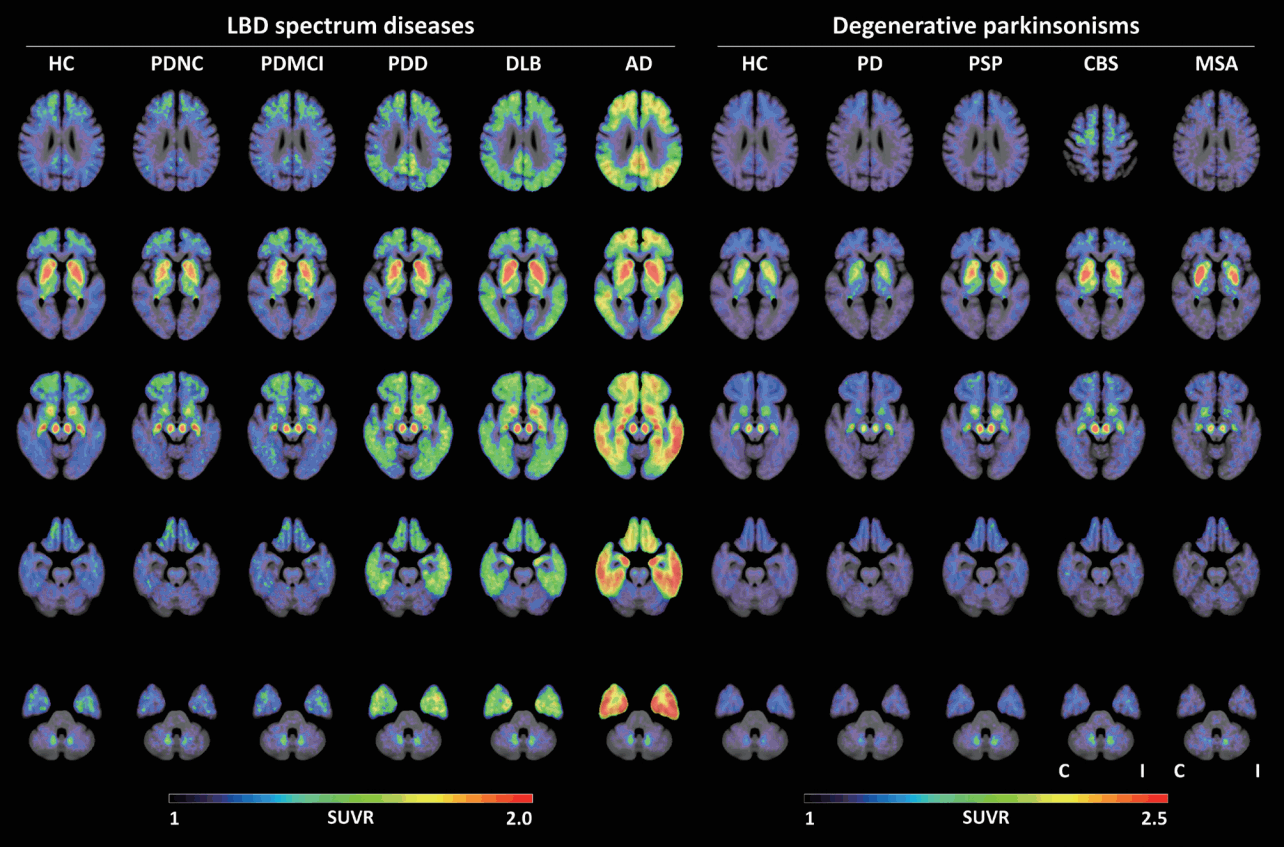
- In summary, the cortical tau burden observed in the 18F-flortaucipir PET study increases within the LBD spectrum (Figure 3). DLB patients exhibit the greatest tau burden, with distribution patterns distinct from AD. Cortical Aβ accumulation may play a greater role in pathological tau accumulation than α-synuclein does. The future development of radiotracers targeting α-synuclein will be helpful in investigating the interaction between the three pathological proteins, as well as for the differential diagnosis of LBD.
LEWY BODY DISEASES
- Unlike the 3R and 4R tau isoform found in AD pathology, the 4R tau isoform is associated with PSP [15]. In PSP, central subcortical gray matter structures, such as the globus pallidus, subthalamic nucleus, and substantia nigra, are most vulnerable to the accumulation of pathological tau protein. In addition to these regions, the striatum, pontine nuclei, dentate nucleus, and cerebellar white matter are the second most vulnerable regions. Tau pathology is also frequently found in the frontal gray and white matter, predominantly in the posterior region, while tau accumulation in the parietal cortex occurs in severely affected patients [71,72]. Although the pathological tau burden is most severe in the PSP-Richardson’s syndrome compared to the PSP-parkinsonism and PSP-pure akinesia with gait freezing types, all PSP subtypes commonly feature the prominent involvement of the central subcortical gray matter structures, and the clinical severity of PSP correlates with pathological tau burden [72].
- The first attempt at in vivo PET imaging of pathological tau protein in PSP was performed with 2-(1-6-[(2-18F-fluoroethyl) (methyl) amino]-2-naphthylethylidene) malononitrile (18F-FDDNP) PET, which non-selectively binds to tau, as well as to Aβ [73]. In this study, PSP patients exhibited increased 18F-FDDNP binding primarily in the subcortical regions, including the striatum, thalamus, subthalamic nucleus, midbrain, and cerebellar white matter. However, PSP rating scale (PSPRS) scores correlated only with the binding in the frontal cortex [73].
- Following the development of the tau-selective radiotracers, six 18F-flortaucipir PET studies, including one case report and one 18F-THK5351 PET study, were published in 2017 [25,26,30-32,67,74]. All of these studies commonly found highly increased radiotracer binding in the globus pallidus and midbrain relative to controls. Five 18F-flortaucipir PET studies additionally found increased binding in the striatum [25,30-32,67], and four studies additionally observed increased binding in the cerebellar dentate nucleus [25,30,32,67]. Only one study showed additionally increased 18F-flortaucipir binding in the frontal cortex [32]. No correlation between disease severity measured by the PSPRS scores and radiotracer binding in any regions was found in any of the three studies [25,30,67], while two studies found a weak correlation between the PSPRS scores and binding in the globus pallidus [31], or that in the midbrain, thalamus, dentate nucleus, precentral cortex, supplementary motor area, middle frontal cortex, and inferior frontal cortex (Figures 2 and 3) [32]. It is very interesting to note that PSP patients can be discriminated by the high 18F-flortaucipir binding in the globus pallidus with 93% sensitivity and 100% specificity [25]. A recent large study including 33 PSP patients and 26 PD patients replicated this finding (85% sensitivity and 92% specificity) [67].
- In contrast to the high in vivo 18F-flortaucipir binding and a high amount of hyperphosphorylated tau in the globus pallidus and midbrain, autoradiography studies of postmortem tissues of PSP brains have shown weak binding of 18F-flortaucipir, as with other types of non-AD tauopathies [18,19,23,30]. It is still questionable whether high in vivo 18F-flortaucipir binding in the globus pallidus and midbrain in PSP is true specific binding with a weak affinity or a result of unknown off-target binding.
- Although there has been some variability seen in tau PET radiotracer binding, increased binding in the globus pallidus and midbrain, which are the most vulnerable to tau pathology in PSP, is considered a characteristic tau PET finding in PSP (Figures 2 and 3). Tau PET may, therefore, be helpful for the differential diagnosis of PD and PSP.
PROGRESSIVE SUPRANUCLEAR PALSY
- CBS is a pathologically heterogeneous clinical syndrome characterized by parkinsonism, dystonia, apraxia, alien hand phenomenon, and myoclonus [75-77]. Corticobasal degeneration (CBD) is a pathological diagnosis accounting for almost half of CBS patients [78-82]. Considering the prevalence of other types of non-AD tauopathies in CBS, non-AD tau pathology can be found in over 70% of CBS patients [78-82]. Tau pathology featuring the 4R-isoform is found most prominently in the superior frontal and parietal cortices, as well as the perirolandic areas and their underlying white matter, and subcortical gray matter structures [83,84].
- Excluding three CBS patients who showed asymmetrically increased 18F-flortaucipir binding in the parietotemporal cortex due to AD [27,85], three 18F-flortaucipir PET studies including one pathologically confirmed CBD patient and one 18F-THK5351 study, have been reported [24,27,81,86,87]. One autoradiography study with 3H-THK5351 showed strong 3H-THK5351 binding in the frontal subcortical white matter, especially in the thread pathology [86]. Moreover, binding intensity in the autoradiography results correlated with the extent of tissue tau pathology [86]. In contrast, another autoradiography study with 18F-flortaucipir showed very weak binding in a small part of the basal ganglia in which the greatest tau pathology existed, but antemortem in vivo 18F-flortaucipir PET binding correlated with tau burden, as measured by immunohistochemical stains of postmortem tissue [81].
- The first tau PET study of 18F-THK5351 in five CBS patients revealed highly increased binding to the perirolandic cortical gray matter and underlying white matter, as well as in the basal ganglia, that was predominant in the side contralateral to the clinically more-affected side [86]. Likewise, two subsequent 18F-flortaucipir PET studies supported this finding [24,27]. Interestingly, these 18F-flortaucipir PET studies commonly found a good correlation between the severity of parkinsonian motor deficit and 18F-flortaucipir binding in the internal capsule [27] or the precentral gray matter and underlying white matter [24].
- Tau PET distribution patterns in CBS patients are characterized by increased radiotracer binding predominantly in the motor cortex and the underlying white matter, as well as in the basal ganglia (Figure 3). Although 18F-flortaucipir binding is generally much weaker in CBS compared to AD, cortical or parkinsonian motor deficits may be attributable to tau burden in motor-related cortical gray matter and white matter, and basal ganglia.
CORTICOBASAL SYNDROME
- Glial cytoplasmic inclusion (GCI) containing α-synuclein is a pathological hallmark of MSA, and can, therefore, be considered a synucleinopathy [88]. Although co-localization of tau pathology in GCIs has been reported in some patients with MSA [89-93], tau pathology was found to be very rare in a postmortem study with a large number of MSA patients [94]. Therefore, it may be unlikely that there is increased 18F-flortaucipir binding in the putamen, where GCI pathology is most prominent. However, one 18F-flortaucipir PET study of four consecutive parkinsonian-type MSA patients clearly showed asymmetrically increased 18F-flortaucipir binding in the atrophic posterior putamen, which was more prominent in the side ipsilateral to the greater putaminal atrophy, together with lower uptake of dopamine transporter PET contralateral to the clinically more affected side [95]. Considering the very low prevalence of tau pathology in MSA, it is unlikely that 18F-flortaucipir bound specifically to tau protein co-localized in the atrophic putamen. Instead, the unexpected results could be attributable to unknown off-target binding.
- In 18F-flortaucipir PET, basal ganglial off-target binding is commonly observed even in healthy elderly individuals [18,41,45]. Interestingly, the topography of subcortical nuclei showing 18F-flortaucipir binding is similar to that of iron in the brain, although an autoradiography study failed to find a spatial match within each region [19]. Greater iron content was demonstrated in the putamen of the MSA brains [96-98], an effect that was replicated in quantitative MR imaging studies of brain iron [99,100]. A recent iron-sensitive quantitative magnetic resonance imaging and 18F-flortaucipir PET study showed a direct correlation between age-related increases in basal ganglial iron content and 18F-flortaucipir binding [46]. Therefore, there may be an in vivo interaction between 18F-flortaucipir and iron. Another possible mechanism for this unexpected binding can be explained by off-target binding to the MAO-B expressed by reactive astrocytes, although 18F-flortaucipir binding to MAO-B has not been proven [101]. However, regardless of the nature of the putaminal 18F-flortaucipir binding in MSA, 18F-flortaucipir PET may be useful for the differential diagnosis of parkinsonism due to its binding topography in the basal ganglia (Figure 3).
MULTIPLE SYSTEM ATROPHY
- Although 18F-flortaucipir is the most promising tau-selective radiotracer for imaging various tauopathies among the tau-selective radiotracers already validated by clinical PET studies, it has drawbacks: off-target binding, unstable kinetics, weak affinity to non-AD tau, and possible MAO binding. Nevertheless, 18F-flortaucipir binds in distinct patterns in different degenerative parkinsonisms and is, therefore, a potential imaging biomarker for the differential diagnosis of parkinsonisms. Next generation tau-selective radiotracers without the problems that are common for the first generation radiotracers will be more helpful for the visualization of tau pathology in degenerative parkinsonisms, as well as in AD. Furthermore, tau PET will be a good imaging biomarker for monitoring the response to pathology-targeted immunotherapy in these tauopathies.
CONCLUSIONS
- This study was financially supported by the “Mirae Medical” Faculty Research Assistance Program of Yonsei University College of Medicine (grant number 6-2016-0162) and the Basic Science Research Program through the NRF funded by the Ministry of Science, ICT & Future Planning (2017R1A2B2006694).
Acknowledgments

Figure 2.Different nigral 18F-flortaucipir binding in PD and PSP. A: Compared to the controls, 18F-flortaucipir SUVR values in the substantia nigra were 13% lower in PD patients and 10% higher in PSP patients. B: A demonstration of different nigral 18F-flortaucipir binding in a control subject and patients with PD and PSP. HC: healthy controls, PD: Parkinson’s disease, PSP: progressive supranuclear palsy, SUVR: standardized uptake value ratio.

Figure 3.Group-averaged 18F-flortaucipir PET images in various degenerative parkinsonisms. In LBD, 18F-flortaucipir PET shows an increasing pattern of cortical binding with the advancement of the disease. In addition, different degenerative parkinsonisms show distinct patterns of 18F-flortaucipir binding; compared to the controls, lower binding has been observed in the substantia nigra in PD, in contrast to higher binding in PSP, as well as higher binding in the globus pallidus and dentate nucleus in PSP, asymmetrically increased binding in the basal ganglia, substantia nigra and white matter underlying the motor cortex in CBS, and asymmetrically increased binding in the putamen in MSA. Color bars represent SUVR values. LBD: Lewy body diseases, HC: healthy controls, PDNC: Parkinson’s disease with normal cognition, PDMCI: Parkinson’s disease with mild cognitive impairment, PDD: Parkinson’s disease with dementia, DLB: dementia with Lewy bodies, AD: Alzheimer’s disease, PSP: progressive supranuclear palsy, CBS: corticobasal syndrome, MSA: multiple system atrophy, C/I: contralateral or ipsilateral to the clinically more affected side, SUVR: standardized uptake value ratio, PET: positron emission tomography.

- 1. Hatashita S, Yamasaki H, Suzuki Y, Tanaka K, Wakebe D, Hayakawa H. [18F]Flutemetamol amyloid-beta PET imaging compared with [11C]PIB across the spectrum of Alzheimer’s disease. Eur J Nucl Med Mol Imaging 2014;41:290–300.ArticlePubMed
- 2. Landau SM, Breault C, Joshi AD, Pontecorvo M, Mathis CA, Jagust WJ, et al. Amyloid-β imaging with Pittsburgh compound B and florbetapir: comparing radiotracers and quantification methods. J Nucl Med 2013;54:70–77.ArticlePubMed
- 3. Rowe CC, Pejoska S, Mulligan RS, Jones G, Chan JG, Svensson S, et al. Head-to-head comparison of 11C-PiB and 18F-AZD4694 (NAV4694) for β-amyloid imaging in aging and dementia. J Nucl Med 2013;54:880–886.ArticlePubMed
- 4. Villemagne VL, Mulligan RS, Pejoska S, Ong K, Jones G, O’Keefe G, et al. Comparison of 11C-PiB and 18F-florbetaben for Aβ imaging in ageing and Alzheimer’s disease. Eur J Nucl Med Mol Imaging 2012;39:983–989.ArticlePubMed
- 5. Driscoll I, Troncoso JC, Rudow G, Sojkova J, Pletnikova O, Zhou Y, et al. Correspondence between in vivo (11)C-PiB-PET amyloid imaging and postmortem, regionmatched assessment of plaques. Acta Neuropathol 2012;124:823–831.ArticlePubMedPMC
- 6. Sojkova J, Driscoll I, Iacono D, Zhou Y, Codispoti KE, Kraut MA, et al. In vivo fibrillar β-amyloid detected using [11C]PiB positron emission tomography and neuropathologic assessment in older adults. Arch Neurol 2011;68:232–240.ArticlePubMedPMC
- 7. Kemppainen NM, Aalto S, Wilson IA, Någren K, Helin S, Brück A, et al. PET amyloid ligand [11C]PIB uptake is increased in mild cognitive impairment. Neurology 2007;68:1603–1606.ArticlePubMed
- 8. Lowe VJ, Kemp BJ, Jack CR Jr, Senjem M, Weigand S, Shiung M, et al. Comparison of 18F-FDG and PiB PET in cognitive impairment. J Nucl Med 2009;50:878–886.ArticlePubMedPMC
- 9. Jack CR Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, et al. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 2013;12:207–216.ArticlePubMedPMC
- 10. Jack CR Jr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: implications for sequence of pathological events in Alzheimer’s disease. Brain 2009;132:1355–1365.ArticlePubMedPMCPDF
- 11. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–259.ArticlePubMedPDF
- 12. Murray ME, Lowe VJ, Graff-Radford NR, Liesinger AM, Cannon A, Przybelski SA, et al. Clinicopathologic and 11C-Pittsburgh compound B implications of Thal amyloid phase across the Alzheimer’s disease spectrum. Brain 2015;138:1370–1381.ArticlePubMedPMCPDF
- 13. Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 1992;42:631–639.ArticlePubMed
- 14. Bierer LM, Hof PR, Purohit DP, Carlin L, Schmeidler J, Davis KL, et al. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer’s disease. Arch Neurol 1995;52:81–88.ArticlePubMed
- 15. Villemagne VL, Fodero-Tavoletti MT, Masters CL, Rowe CC. Tau imaging: early progress and future directions. Lancet Neurol 2015;14:114–124.ArticlePubMed
- 16. Fodero-Tavoletti MT, Okamura N, Furumoto S, Mulligan RS, Connor AR, McLean CA, et al. 18F-THK523: a novel in vivo tau imaging ligand for Alzheimer’s disease. Brain 2011;134:1089–1100.ArticlePubMedPDF
- 17. Villemagne VL, Furumoto S, Fodero-Tavoletti MT, Mulligan RS, Hodges J, Harada R, et al. In vivo evaluation of a novel tau imaging tracer for Alzheimer’s disease. Eur J Nucl Med Mol Imaging 2014;41:816–826.ArticlePubMedPDF
- 18. Marquié M, Normandin MD, Vanderburg CR, Costantino IM, Bien EA, Rycyna LG, et al. Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol 2015;78:787–800.ArticlePubMedPMC
- 19. Lowe VJ, Curran G, Fang P, Liesinger AM, Josephs KA, Parisi JE, et al. An autoradiographic evaluation of AV-1451 Tau PET in dementia. Acta Neuropathol Commun 2016;4:58.ArticlePubMedPMC
- 20. Cho H, Choi JY, Hwang MS, Kim YJ, Lee HM, Lee HS, et al. In vivo cortical spreading pattern of tau and amyloid in the Alzheimer disease spectrum. Ann Neurol 2016;80:247–258.ArticlePubMed
- 21. Cho H, Choi JY, Hwang MS, Lee JH, Kim YJ, Lee HM, et al. Tau PET in Alzheimer disease and mild cognitive impairment. Neurology 2016;87:375–383.ArticlePubMed
- 22. Johnson KA, Schultz A, Betensky RA, Becker JA, Sepulcre J, Rentz D, et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol 2016;79:110–119.ArticlePubMed
- 23. Marquié M, Normandin MD, Meltzer AC, Siao Tick Chong M, Andrea NV, Antón-Fernández A, et al. Pathological correlations of [F-18]-AV-1451 imaging in non-alzheimer tauopathies. Ann Neurol 2017;81:117–128.ArticlePubMedPMC
- 24. Cho H, Baek MS, Choi JY, Lee SH, Kim JS, Ryu YH, et al. 18F-AV-1451 binds to motor-related subcortical gray and white matter in corticobasal syndrome. Neurology 2017;89:1170–1178.ArticlePubMed
- 25. Cho H, Choi JY, Hwang MS, Lee SH, Ryu YH, Lee MS, et al. Subcortical 18 F-AV-1451 binding patterns in progressive supranuclear palsy. Mov Disord 2017;32:134–140.ArticlePubMed
- 26. Hammes J, Bischof GN, Giehl K, Faber J, Drzezga A, Klockgether T, et al. Elevated in vivo [18F]-AV-1451 uptake in a patient with progressive supranuclear palsy. Mov Disord 2017;32:170–171.ArticlePubMed
- 27. Smith R, Schöll M, Widner H, van Westen D, Svenningsson P, Hägerström D, et al. In vivo retention of 18F-AV-1451 in corticobasal syndrome. Neurology 2017;89:845–853.ArticlePubMedPMC
- 28. Spina S, Schonhaut DR, Boeve BF, Seeley WW, Ossenkoppele R, O’Neil JP, et al. Frontotemporal dementia with the V337M MAPT mutation: Tau-PET and pathology correlations. Neurology 2017;88:758–766.ArticlePubMedPMC
- 29. Smith R, Puschmann A, Schöll M, Ohlsson T, van Swieten J, Honer M, et al. 18F-AV-1451 tau PET imaging correlates strongly with tau neuropathology in MAPT mutation carriers. Brain 2016;139:2372–2379.ArticlePubMedPMCPDF
- 30. Passamonti L, Vázquez Rodríguez P, Hong YT, Allinson KS, Williamson D, Borchert RJ, et al. 18F-AV-1451 positron emission tomography in Alzheimer’s disease and progressive supranuclear palsy. Brain 2017;140:781–791.ArticlePubMedPMC
- 31. Smith R, Schain M, Nilsson C, Strandberg O, Olsson T, Hägerström D, et al. Increased basal ganglia binding of 18 F-AV-1451 in patients with progressive supranuclear palsy. Mov Disord 2017;32:108–114.ArticlePubMed
- 32. Whitwell JL, Lowe VJ, Tosakulwong N, Weigand SD, Senjem ML, Schwarz CG, et al. [18F]AV-1451 tau positron emission tomography in progressive supranuclear palsy. Mov Disord 2017;32:124–133.ArticlePubMed
- 33. Harada R, Okamura N, Furumoto S, Furukawa K, Ishiki A, Tomita N, et al. 18F-THK5351: a novel PET radiotracer for imaging neurofibrillary pathology in Alzheimer disease. J Nucl Med 2016;57:208–214.ArticlePubMed
- 34. Ng KP, Pascoal TA, Mathotaarachchi S, Therriault J, Kang MS, Shin M, et al. Monoamine oxidase B inhibitor, selegiline, reduces 18F-THK5351 uptake in the human brain. Alzheimers Res Ther 2017;9:25.ArticlePubMedPMCPDF
- 35. Jossan SS, Gillberg PG, d’Argy R, Aquilonius SM, Långström B, Halldin C, et al. Quantitative localization of human brain monoamine oxidase B by large section autoradiography using L-[3H]deprenyl. Brain Res 1991;547:69–76.ArticlePubMed
- 36. Fowler JS, MacGregor RR, Wolf AP, Arnett CD, Dewey SL, Schlyer D, et al. Mapping human brain monoamine oxidase A and B with 11C-labeled suicide inactivators and PET. Science 1987;235:481–485.ArticlePubMed
- 37. Fowler JS, Volkow ND, Wang GJ, Logan J, Pappas N, Shea C, et al. Age-related increases in brain monoamine oxidase B in living healthy human subjects. Neurobiol Aging 1997;18:431–435.ArticlePubMed
- 38. Ginovart N, Meyer JH, Boovariwala A, Hussey D, Rabiner EA, Houle S, et al. Positron emission tomography quantification of [11C]-harmine binding to monoamine oxidase-a in the human brain. J Cereb Blood Flow Metab 2006;26:330–344.ArticlePubMed
- 39. Xia CF, Arteaga J, Chen G, Gangadharmath U, Gomez LF, Kasi D, et al. [(18)F]T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimers Dement 2013;9:666–676.ArticlePubMed
- 40. Chien DT, Szardenings AK, Bahri S, Walsh JC, Mu F, Xia C, et al. Early clinical PET imaging results with the novel PHF-tau radioligand [F18]-T808. J Alzheimers Dis 2014;38:171–184.ArticlePubMed
- 41. Chien DT, Bahri S, Szardenings AK, Walsh JC, Mu F, Su MY, et al. Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807. J Alzheimers Dis 2013;34:457–468.ArticlePubMed
- 42. Baker SL, Lockhart SN, Price JC, He M, Huesman RH, Schonhaut D, et al. Reference tissue-based kinetic evaluation of 18F-AV-1451 for tau imaging. J Nucl Med 2017;58:332–338.ArticlePubMedPMC
- 43. Shcherbinin S, Schwarz AJ, Joshi A, Navitsky M, Flitter M, Shankle WR, et al. Kinetics of the tau PET tracer 18F-AV-1451 (T807) in subjects with normal cognitive function, mild cognitive impairment, and Alzheimer disease. J Nucl Med 2016;57:1535–1542.ArticlePubMed
- 44. Wooten DW, Guehl NJ, Verwer EE, Shoup TM, Yokell DL, Zubcevik N, et al. Pharmacokinetic evaluation of the tau PET radiotracer 18F-T807 (18F-AV-1451) in human subjects. J Nucl Med 2017;58:484–491.ArticlePubMedPMC
- 45. Sander K, Lashley T, Gami P, Gendron T, Lythgoe MF, Rohrer JD, et al. Characterization of tau positron emission tomography tracer [18F]AV-1451 binding to postmortem tissue in Alzheimer’s disease, primary tauopathies, and other dementias. Alzheimers Dement 2016;12:1116–1124.ArticlePubMed
- 46. Choi JY, Cho H, Ahn SJ, Lee JH, Ryu YH, Lee MS, et al. Off-target (18)F-AV-1451 binding in the basal ganglia correlates with age-related iron accumulation. J Nucl Med 2018;59:117–120.ArticlePubMed
- 47. Ikonomovic MD, Abrahamson EE, Price JC, Mathis CA, Klunk WE. [F-18]AV-1451 positron emission tomography retention in choroid plexus: more than “off-target” binding. Ann Neurol 2016;80:307–308.ArticlePubMedPMC
- 48. Hostetler ED, Walji AM, Zeng Z, Miller P, Bennacef I, Salinas C, et al. Preclinical characterization of 18F-MK-6240, a promising PET tracer for in vivo quantification of human neurofibrillary tangles. J Nucl Med 2016;57:1599–1606.ArticlePubMed
- 49. Saint-Aubert L, Lemoine L, Chiotis K, Leuzy A, Rodriguez-Vieitez E, Nordberg A. Tau PET imaging: present and future directions. Mol Neurodegener 2017;12:19.ArticlePubMedPMCPDF
- 50. Lohith T, Bennacef I, Sur C, Declercq R, Serdons K, Bormans G, et al. Quantification of [18F]MK-6240, a new PET tracer targeting human neurofibrillary tangles (NFTs) in brain of healthy elderly and subjects with Alzheimer’s disease. J Nucl Med 2017;58:277.
- 51. Maruyama M, Shimada H, Suhara T, Shinotoh H, Ji B, Maeda J, et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron 2013;79:1094–1108.ArticlePubMed
- 52. Ono M, Sahara N, Kumata K, Ji B, Ni R, Koga S, et al. Distinct binding of PET ligands PBB3 and AV-1451 to tau fibril strains in neurodegenerative tauopathies. Brain 2017;140:764–780.ArticlePubMedPMC
- 53. Hashimoto H, Kawamura K, Igarashi N, Takei M, Fujishiro T, Aihara Y, et al. Radiosynthesis, photoisomerization, biodistribution, and metabolite analysis of 11C-PBB3 as a clinically useful PET probe for imaging of tau pathology. J Nucl Med 2014;55:1532–1538.ArticlePubMed
- 54. Kimura Y, Ichise M, Ito H, Shimada H, Ikoma Y, Seki C, et al. PET quantification of tau pathology in human brain with 11C-PBB3. J Nucl Med 2015;56:1359–1365.ArticlePubMed
- 55. Gomperts SN. Imaging the role of amyloid in PD dementia and dementia with Lewy bodies. Curr Neurol Neurosci Rep 2014;14:472.ArticlePubMedPDF
- 56. Lippa CF, Duda JE, Grossman M, Hurtig HI, Aarsland D, Boeve BF, et al. DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology 2007;68:812–819.ArticlePubMed
- 57. Ballard C, Ziabreva I, Perry R, Larsen JP, O’Brien J, McKeith I, et al. Differences in neuropathologic characteristics across the Lewy body dementia spectrum. Neurology 2006;67:1931–1934.ArticlePubMed
- 58. Jellinger KA, Attems J. Prevalence and impact of vascular and Alzheimer pathologies in Lewy body disease. Acta Neuropathol 2008;115:427–436.ArticlePubMedPDF
- 59. Burack MA, Hartlein J, Flores HP, Taylor-Reinwald L, Perlmutter JS, Cairns NJ. In vivo amyloid imaging in autopsy-confirmed Parkinson disease with dementia. Neurology 2010;74:77–84.ArticlePubMedPMC
- 60. Foster ER, Campbell MC, Burack MA, Hartlein J, Flores HP, Cairns NJ, et al. Amyloid imaging of Lewy body-associated disorders. Mov Disord 2010;25:2516–2523.ArticlePubMedPMC
- 61. Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, et al. Imaging beta-amyloid burden in aging and dementia. Neurology 2007;68:1718–1725.ArticlePubMed
- 62. Edison P, Rowe CC, Rinne JO, Ng S, Ahmed I, Kemppainen N, et al. Amyloid load in Parkinson’s disease dementia and Lewy body dementia measured with [11C]PIB positron emission tomography. J Neurol Neurosurg Psychiatry 2008;79:1331–1338.ArticlePubMed
- 63. Maetzler W, Liepelt I, Reimold M, Reischl G, Solbach C, Becker C, et al. Cortical PIB binding in Lewy body disease is associated with Alzheimer-like characteristics. Neurobiol Dis 2009;34:107–112.ArticlePubMed
- 64. Gomperts SN, Rentz DM, Moran E, Becker JA, Locascio JJ, Klunk WE, et al. Imaging amyloid deposition in Lewy body diseases. Neurology 2008;71:903–910.ArticlePubMedPMC
- 65. Petrou M, Dwamena BA, Foerster BR, MacEachern MP, Bohnen NI, Müller ML, et al. Amyloid deposition in Parkinson’s disease and cognitive impairment: a systematic review. Mov Disord 2015;30:928–935.ArticlePubMedPMC
- 66. Hansen AK, Knudsen K, Lillethorup TP, Landau AM, Parbo P, Fedorova T, et al. In vivo imaging of neuromelanin in Parkinson’s disease using 18F-AV-1451 PET. Brain 2016;139:2039–2049.ArticlePubMedPDF
- 67. Schonhaut DR, McMillan CT, Spina S, Dickerson BC, Siderowf A, Devous MD, et al. 18F-flortaucipir tau positron emission tomography distinguishes established progressive supranuclear palsy from controls and Parkinson disease: a multicenter study. Ann Neurol 2017;82:622–634.ArticlePubMedPMC
- 68. Gomperts SN, Locascio JJ, Makaretz SJ, Schultz A, Caso C, Vasdev N, et al. Tau positron emission tomographic imaging in the Lewy body diseases. JAMA Neurol 2016;73:1334–1341.ArticlePubMedPMC
- 69. Lee SH, Cho H, Choi JY, Lee JH, Ryu YH, Lee MS, et al. Distinct patterns of amyloid-dependent tau accumulation in Lewy body diseases. Mov Disord 2017;Nov. 23. [Epub]. https://doi.org/10.1002/mds.27252. Article
- 70. Kantarci K, Lowe VJ, Boeve BF, Senjem ML, Tosakulwong N, Lesnick TG, et al. AV-1451 tau and β-amyloid positron emission tomography imaging in dementia with Lewy bodies. Ann Neurol 2017;81:58–67.ArticlePubMed
- 71. Williams DR, de Silva R, Paviour DC, Pittman A, Watt HC, Kilford L, et al. Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson’s syndrome and PSP-parkinsonism. Brain 2005;128:1247–1258.ArticlePubMedPDF
- 72. Williams DR, Holton JL, Strand C, Pittman A, de Silva R, Lees AJ, et al. Pathological tau burden and distribution distinguishes progressive supranuclear palsy-parkinsonism from Richardson’s syndrome. Brain 2007;130:1566–1576.ArticlePubMedPDF
- 73. Kepe V, Bordelon Y, Boxer A, Huang SC, Liu J, Thiede FC, et al. PET imaging of neuropathology in tauopathies: progressive supranuclear palsy. J Alzheimers Dis 2013;36:145–153.ArticlePubMedPMC
- 74. Ishiki A, Harada R, Okamura N, Tomita N, Rowe CC, Villemagne VL, et al. Tau imaging with [18 F]THK-5351 in progressive supranuclear palsy. Eur J Neurol 2017;24:130–136.ArticlePubMed
- 75. Boeve BF, Maraganore DM, Parisi JE, Ahlskog JE, Graff- Radford N, Caselli RJ, et al. Pathologic heterogeneity in clinically diagnosed corticobasal degeneration. Neurology 1999;53:795–800.ArticlePubMed
- 76. Kouri N, Whitwell JL, Josephs KA, Rademakers R, Dickson DW. Corticobasal degeneration: a pathologically distinct 4R tauopathy. Nat Rev Neurol 2011;7:263–272.ArticlePubMedPMCPDF
- 77. Mathew R, Bak TH, Hodges JR. Diagnostic criteria for corticobasal syndrome: a comparative study. J Neurol Neurosurg Psychiatry 2012;83:405–410.ArticlePubMed
- 78. McMonagle P, Blair M, Kertesz A. Corticobasal degeneration and progressive aphasia. Neurology 2006;67:1444–1451.ArticlePubMed
- 79. Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol 2003;54 Suppl 5:S15–S19.ArticlePubMed
- 80. Lee SE, Rabinovici GD, Mayo MC, Wilson SM, Seeley WW, DeArmond SJ, et al. Clinicopathological correlations in corticobasal degeneration. Ann Neurol 2011;70:327–340.ArticlePubMedPMC
- 81. Josephs KA, Whitwell JL, Tacik P, Duffy JR, Senjem ML, Tosakulwong N, et al. [18F]AV-1451 tau-PET uptake does correlate with quantitatively measured 4R-tau burden in autopsy-confirmed corticobasal degeneration. Acta Neuropathol 2016;132:931–933.ArticlePubMedPMCPDF
- 82. Ling H, O’Sullivan SS, Holton JL, Revesz T, Massey LA, Williams DR, et al. Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain 2010;133:2045–2057.ArticlePubMedPDF
- 83. Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K, et al. Office of rare diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol 2002;61:935–946.ArticlePubMed
- 84. Ling H, Kovacs GG, Vonsattel JP, Davey K, Mok KY, Hardy J, et al. Astrogliopathy predominates the earliest stage of corticobasal degeneration pathology. Brain 2016;139:3237–3252.ArticlePubMedPDF
- 85. Ossenkoppele R, Schonhaut DR, Schöll M, Lockhart SN, Ayakta N, Baker SL, et al. Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain 2016;139:1551–1567.ArticlePubMedPMCPDF
- 86. Kikuchi A, Okamura N, Hasegawa T, Harada R, Watanuki S, Funaki Y, et al. In vivo visualization of tau deposits in corticobasal syndrome by 18F-THK5351 PET. Neurology 2016;87:2309–2316.ArticlePubMedPMC
- 87. Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol 2004;55:306–319.ArticlePubMed
- 88. Jellinger KA. Neuropathology of multiple system atrophy: new thoughts about pathogenesis. Mov Disord 2014;29:1720–1741.ArticlePubMed
- 89. Nagaishi M, Yokoo H, Nakazato Y. Tau-positive glial cytoplasmic granules in multiple system atrophy. Neuropathology 2011;31:299–305.ArticlePubMed
- 90. Piao YS, Hayashi S, Hasegawa M, Wakabayashi K, Yamada M, Yoshimoto M, et al. Co-localization of alpha-synuclein and phosphorylated tau in neuronal and glial cytoplasmic inclusions in a patient with multiple system atrophy of long duration. Acta Neuropathol 2001;101:285–293.ArticlePubMed
- 91. Takanashi M, Ohta S, Matsuoka S, Mori H, Mizuno Y. Mixed multiple system atrophy and progressive supranuclear palsy: a clinical and pathological report of one case. Acta Neuropathol 2002;103:82–87.ArticlePubMedPDF
- 92. Takeda A, Arai N, Komori T, Iseki E, Kato S, Oda M. Tau immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci Lett 1997;234:63–66.ArticlePubMed
- 93. Uchikado H, DelleDonne A, Uitti R, Dickson DW. Coexistence of PSP and MSA: a case report and review of the literature. Acta Neuropathol 2006;111:186–192.ArticlePubMedPDF
- 94. Jellinger K. Unusual tau in MSA. Neuropathology 2012;32:110–111.ArticlePubMed
- 95. Cho H, Choi JY, Lee SH, Ryu YH, Lee MS, Lyoo CH. 18F-AV-1451 binds to putamen in multiple system atrophy. Mov Disord 2017;32:171–173.ArticlePubMed
- 96. Dexter DT, Carayon A, Javoy-Agid F, Agid Y, Wells FR, Daniel SE, et al. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain 1991;114:1953–1975.ArticlePubMedPDF
- 97. Dickson DW, Lin W, Liu WK, Yen SH. Multiple system atrophy: a sporadic synucleinopathy. Brain Pathol 1999;9:721–732.ArticlePubMed
- 98. Kato S, Meshitsuka S, Ohama E, Tanaka J, Llena JF, Hirano A. Increased iron content in the putamen of patients with striatonigral degeneration. Acta Neuropathol 1992;84:328–330.ArticlePubMedPDF
- 99. Lee JH, Han YH, Kang BM, Mun CW, Lee SJ, Baik SK. Quantitative assessment of subcortical atrophy and iron content in progressive supranuclear palsy and parkinsonian variant of multiple system atrophy. J Neurol 2013;260:2094–2101.ArticlePubMedPDF
- 100. Lee JH, Kim TH, Mun CW, Kim TH, Han YH. Progression of subcortical atrophy and iron deposition in multiple system atrophy: a comparison between clinical subtypes. J Neurol 2015;262:1876–1882.ArticlePubMedPDF
- 101. Bevan-Jones WR, Cope TE, Jones PS, Passamonti L, Hong YT, Fryer TD, et al. [18F]AV-1451 binding in vivo mirrors the expected distribution of TDP-43 pathology in the semantic variant of primary progressive aphasia. J Neurol Neurosurg Psychiatry 2017;Sep. 14. [Epub]. https://doi.org/10.1136/jnnp-2017-316402. Article
REFERENCES
Figure & Data
References
Citations
Citations to this article as recorded by 
- Current directions in tau research: Highlights from Tau 2020
Claire Sexton, Heather Snyder, Dirk Beher, Adam L. Boxer, Pat Brannelly, Jean‐Pierre Brion, Luc Buée, Angela M. Cacace, Gaël Chételat, Martin Citron, Sarah L. DeVos, Kristophe Diaz, Howard H. Feldman, Bess Frost, Alison M. Goate, Michael Gold, Bradley Hym
Alzheimer's & Dementia.2022; 18(5): 988. CrossRef - 18F‐Florzolotau Tau Positron Emission Tomography Imaging in Patients with Multiple System Atrophy–Parkinsonian Subtype
Feng‐Tao Liu, Xin‐Yi Li, Jia‐Ying Lu, Ping Wu, Ling Li, Xiao‐Niu Liang, Zi‐Zhao Ju, Fang‐Yang Jiao, Ming‐Jia Chen, Jing‐Jie Ge, Yi‐Min Sun, Jian‐Jun Wu, Tzu‐Chen Yen, Jian‐Feng Luo, Chuantao Zuo, Jian Wang
Movement Disorders.2022; 37(9): 1915. CrossRef - Imaging pathological tau in atypical parkinsonisms: A review
Anastassia M. Mena, Antonio P. Strafella
Clinical Parkinsonism & Related Disorders.2022; 7: 100155. CrossRef - Integrated 18F-T807 Tau PET, Structural MRI, and Plasma Tau in Tauopathy Neurodegenerative Disorders
Cheng-Hsuan Li, Ta-Fu Chen, Ming-Jang Chiu, Ruoh-Fang Yen, Ming-Chieh Shih, Chin-Hsien Lin
Frontiers in Aging Neuroscience.2021;[Epub] CrossRef - Dual-Phase 18F-FP-CIT PET in Corticobasal Syndrome
Je Hong Min, Dong Gyu Park, Jung Han Yoon, Young Sil An
Clinical Nuclear Medicine.2019; 44(1): e49. CrossRef - Tau Positron-Emission Tomography in Former National Football League Players
Robert A. Stern, Charles H. Adler, Kewei Chen, Michael Navitsky, Ji Luo, David W. Dodick, Michael L. Alosco, Yorghos Tripodis, Dhruman D. Goradia, Brett Martin, Diego Mastroeni, Nathan G. Fritts, Johnny Jarnagin, Michael D. Devous, Mark A. Mintun, Michael
New England Journal of Medicine.2019; 380(18): 1716. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite