 E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 16(2); 2023 > Article
-
Review Article
Adult-Onset Genetic Leukoencephalopathies With Movement Disorders -
Mu-Hui Fu1,2*

 , Yung-Yee Chang1,2*
, Yung-Yee Chang1,2* -
Journal of Movement Disorders 2023;16(2):115-132.
DOI: https://doi.org/10.14802/jmd.22127
Published online: March 7, 2023
1Department of Neurology, Kaohsiung Chang Gung Memorial Hospital, Chang Gung University College of Medicine, Kaohsiung, Taiwan
2Center for Parkinson’s Disease, Kaohsiung Chang Gung Memorial Hospital, Kaohsiung, Taiwan
- Corresponding author: Mu-Hui Fu, MD, PhD Department of Neurology, Kaohsiung Chang Gung Memorial Hospital, No. 123, Dapi Rd., Niaosong District, Kaohsiung 83301, Taiwan / Tel: +886-7-7317123-3399 / Fax: +886-7-7317123-3390 / E-mail: kf4089@gmail.com
- Corresponding author: Yung-Yee Chang, MD Department of Neurology, Kaohsiung Chang Gung Memorial Hospital, No. 123, Dapi Rd., Niaosong District, Kaohsiung 83301, Taiwan / Tel: +886-7-7317123-3399 / Fax: +886-7-7317123-3390 / E-mail: changyy7@gmail.com
- *This authors contributed equally to this work.
Copyright © 2023 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 4,030 Views
- 348 Download
ABSTRACT
- Genetic leukoencephalopathies (GLEs) are a group of white matter abnormalities with heterogeneous radiological and phenotypic features. Although these conditions have mostly been described in children, adult-onset cases are increasingly recognized owing to the widespread use of neuroimaging and advances in molecular genetic testing. The disease course is often progressive with a varied spectrum of presentations, trapping neurologists in the dilemma of differential diagnosis. Movement disorders are among the most common symptoms, and their diversity makes diagnosis challenging. In this review, we focus on adult-onset GLEs with movement disorders and offer a step-by-step diagnostic approach by clarifying the phenomenology of movement, advising investigations for acquired causes, describing the clinical and radiological clues to each disease, emphasizing the limitations of advanced molecular testing, and discussing the future application of artificial intelligence. We provide a list summarizing the leukoencephalopathies associated with different categories of movement disorders. In addition to guiding clinicians on how to narrow the list of differential diagnoses with the tools currently available, another aim of this review is to emphasize the inevitable trend toward applying advanced technology in diagnosing these difficult diseases.
- Keywords: Chorea; Dystonia; Leukoencephalopathy; Movement disorders; Parkinsonism; Tremor
- Leukoencephalopathies are disorders that mainly affect the white matter of the brain. The term was initially introduced through a pathological or biochemical analysis of brain tissue but increased in use after the emergence of neuroimaging, especially magnetic resonance imaging (MRI). Widespread hyperintense signal abnormalities in the brain on T2-weighted image (T2WI) and fluid-attenuated inversion-recovery (FLAIR) MRI are the typical radiological features. In the adult population, acquired causes of leukoencephalopathies, such as vascular disorders, neuroinflammation, infections, neoplasms, and traumatic or toxic causes, are more common than genetic etiologies. However, with advances in molecular genetic testing, a growing number of genetic leukoencephalopathies (GLEs) have been identified [1].
- Another confusing term is “leukodystrophies”, which represent a group of inherited disorders that selectively impair the development or maintenance of central nervous system (CNS) myelin. The neuropathological hallmark of leukodystrophies is hypomyelination or demyelination, with CNS glial cells being the most frequently affected cell types. The terms “leukodystrophy” and “GLE” are sometimes used interchangeably, but there has been a consensus regarding how these terms are defined and applied [2]. Notably, leukodystrophies are GLEs, but not all GLEs are classified as leukodystrophies. Typically, GLEs present in early infancy or childhood, but many of them develop phenotypes later in adulthood [3,4]. Raising awareness while approaching adult patients is urgent because some of them are treatable or open to intervention.
- The constellation of neurological manifestations in this group of diseases is highly variable, consisting of cognitive impairment, behavioral changes, vision/hearing problems, movement disorders, gait disturbance, upper motor neuron signs, seizures, autonomic dysfunction, and peripheral neuropathy. Moreover, GLEs have significantly heterogeneous extraneurological features, as well as neuroimaging findings. The Online Mendelian Inheritance in Man (OMIM) full-text entry “leukoencephalopathy” or “leukodystrophy” reports 353 associated genes (21 July, 2022) [5]. Such heterogeneity in the pathological mechanisms, clinical manifestations, radiological features, and loose phenotype-genotype correlations make diagnosis challenging. Thus, the application of broad-spectrum next-generation sequencing (NGS), such as whole-exome or whole-genome genetic testing, appears reasonable. Several authors have reviewed these diseases and summarized the approaching methods according to their experience [6-9]; however, research focusing on movement disorder-relevant leukoencephalopathy has been lacking.
- The purpose of this review is to provide a practical approach for the diagnosis of GLEs with movement disorders using the current available information. Several common and important diseases are introduced, but to encompass all diseases and their atypical presentations is beyond the scope of this review.
INTRODUCTION
- Step 1: Clarifying the movement disorders
- Movement disorders can be classified into three categories: increased movement (hyperkinetic disorder), decreased movement (hypokinetic disorder), and ataxia. Hyperkinesias are characterized by abnormal, uncontrollable, and unwanted excess movements. In contrast, hypokinesias are a lack of voluntary movements (akinesia), accompanied by slow movements (bradykinesia), inappropriately reduced amplitude of movements (hypokinesia), and stiffness or increased muscle tone (rigidity). Hyperkinetic movement disorders comprise dystonia, tremor, chorea, myoclonus, and stereotypies [10,11]. The terms akinesia, bradykinesia, and hypokinesia are used interchangeably to describe the basic motor abnormalities seen in parkinsonism. Ataxia is not a hypo- or hyperkinetic movement disorder; however, it manifests as loss of coordination, causing impaired limb muscle control, gait disturbances, eye movement disorders, and dysarthria.
- When approaching a patient with movement disorder, we recommend the “two-axes” classification system proposed in the clinical approach to dystonia from the International Parkinson and Movement Disorder Society [12]. In Axis I, four dimensions of clinical manifestations should be addressed, including the affected body region, age at onset, temporal pattern, and any associated features (pay attention to ocular manifestations, tendinous xanthoma, skin color, diarrhea, neuropathy/myopathy). Axis II focuses on etiology and should obtain two key pieces of information: identifiable underlying neuropathology and pattern of inheritance. Abnormal movement can appear in isolation or in variable combinations, and the predominant involuntary movements of certain GLEs are summarized in Figure 1.
- Step 2: Excluding common acquired leukoencephalopathies
- As depicted by Ahmed et al. [13], excluding possible secondary causes of leukoencephalopathy is the first diagnostic priority for clinicians. These causes include the following.
- - History, neurological/physical examinations: Exposure to drugs/addictive substances/immunosuppressants, alcohol use disorders, fasting/malabsorption/abdominal operation (e.g., prior gastrectomy), chemotherapy (especially 5-fluorouracil and methotrexate [14])/radiotherapy, blood pressure surge/shock event, eclampsia, signs of venous sinus thrombosis, prior electrolyte imbalance and related treatment, hypoxia, and sleep disturbances.
- - Blood tests: Sugar, electrolytes, hepatic/renal function, thyroid function, autoimmune diseases (especially systemic lupus erythematosus), cortisol, vitamin B12, folate, human immunodeficiency virus (HIV), syphilis, hepatitis B/C, copper/ceruloplasmin, angiotensin converting enzyme, anti-aquaporin-4 (AQP-4) antibody, and anti-myelin oligodendrocyte glycoprotein (anti-MOG) antibody.
- - Cerebrospinal fluid (CSF) studies: Any sign of inflammation/infection, oligoclonal bands, John Cunningham (JC) virus survey if there is a history of immunosuppression, and autoimmune/paraneoplastic encephalitis panel.
- - Nerve conduction studies
- - Evoked potentials
- - MRI with gadolinium enhancement: CNS lymphoma, gliomatosis cerebri, multiple sclerosis, neuromyelitis optica spectrum disorders, anti-MOG associated encephalomyelitis, and acute disseminated encephalomyelitis should be excluded as much as possible based on the Axis I information (clinical manifestations, temporal pattern) [15].
- Step 3: MRI-centered approach—MRI characteristics of each GLE
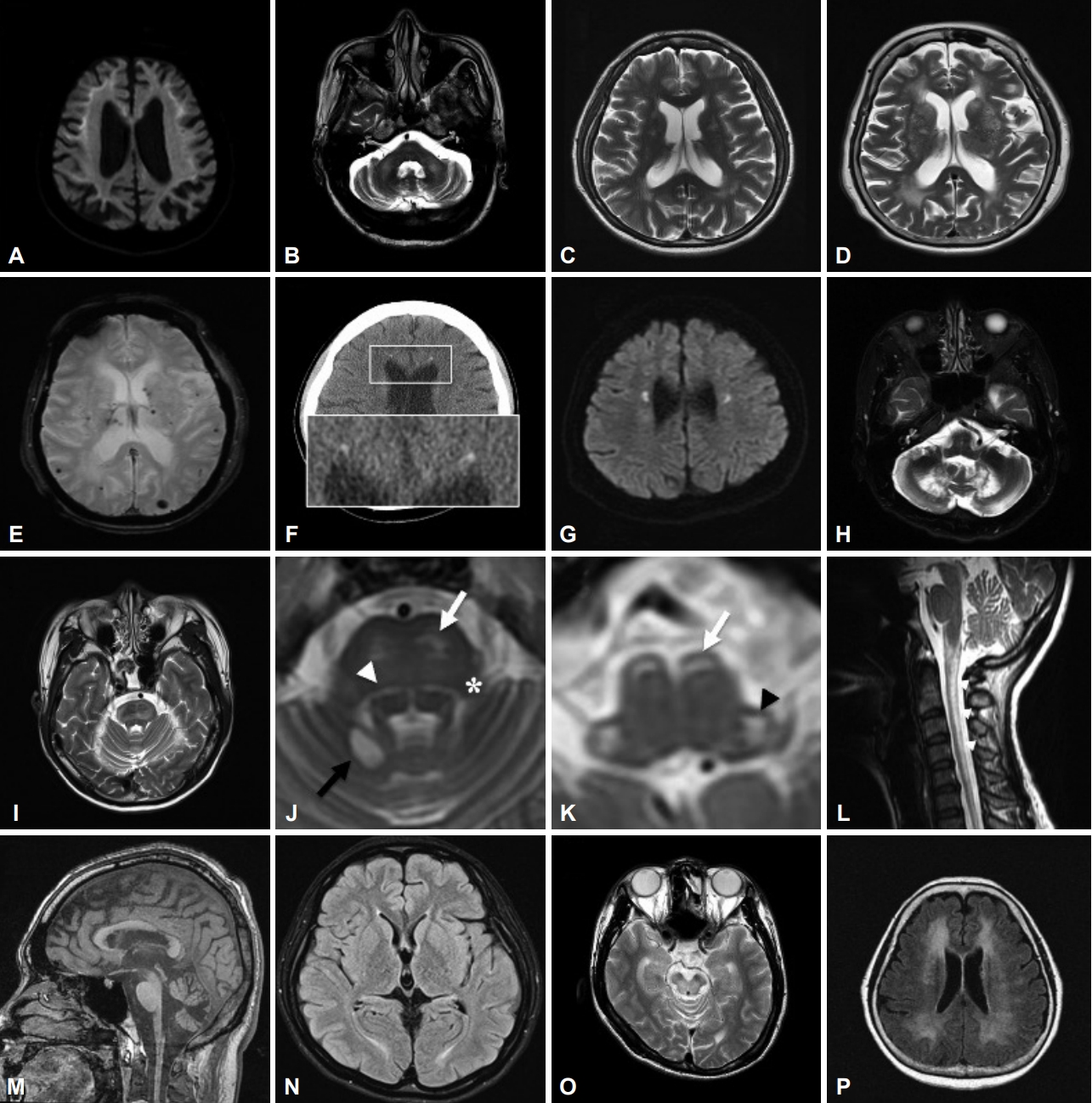
- MRI is a pivotal tool in the diagnostic workup of patients with leukoencephalopathies. The interpretation of imaging should be based on the Axis I assessment, although sometimes it provides diagnostic clues even before complete collection of this information. T2WI and FLAIR MRI are the most useful sequences to determine white matter involvement. In addition, T1-weighted imaging (T1WI) can assist in certain heavy metal accumulation diseases, and diffusion-weighted imaging (DWI) can provide additional information, such as the high-intensity signals along the U-fibers in neuronal intranuclear inclusion disease (NIID) and small deep white matter lesions in CSF1R-related leukoencephalopathy. Figure 2 and Table 1 provide the major radiological characteristics of certain GLEs, and approaching patients with such a “pattern recognition” method allows clinicians to arrange more tailored investigations. Serial MRI scans at an interval of 6–12 months are often needed. Sometimes more than one pattern can be recognized during the disease course of the same patient. Several distinctive features can help to narrow the differential diagnosis under such conditions.
- However, we must bear in mind that there are always exceptions, and white matter involvement is usually too extensive to distinguish or too trivial to notice. Thus, we suggest commencing serial biochemical screening for metabolic leukoencephalopathies if MRI pattern recognition fails. Since X-linked adrenoleukodystrophy (ALD) is the most common peroxisomal disorder, very-long-chain fatty acid (VLCFA) measurement for the diagnosis of it should be performed early, especially in male patients presenting with gait problems [16]. Other tests include white cell enzymes for metachromatic leukodystrophy (MLD), Krabbe disease, GM1/GM2 gangliosidosis; blood lactate, pyruvate, amino acids/urine organic acids for mitochondrial diseases; and plasma amino acid profile and total homocysteine for methylenetetrahydrofolate reductase deficiency and homocystinuria [7,13,17].
- Step 4: Check for possible repeat expansion diseases/mitochondrial DNA (mtDNA) mutation diseases
- If biochemical/metabolic testing is unrevealing, unavailable, or slow in processing, and the patient’s clinical manifestations raise a strong suspicion of certain diseases, a gene-targeted screening panel for repeat expansion diseases or mtDNA is indicated. The discovery of these diseases overrules the conventional rules of Mendelian inheritance since these mutations are unstable and often change size within generations [18]. Most currently available NGS platforms are unable to genotype large and/or complex repeat expansions; thus, applying an appropriate methodology is pivotal to overcome this limitation. Owing to widely varying phenotypes and limited detection tools, it is conceivable that the list of these disorders will continue to grow in the future. Here, we introduce several important repeat expansion disorders with white matter abnormalities and movement disorders.
- The clinical features of NIID are heterogeneous, comprising cognitive impairment, behavior changes, ataxia, muscle weakness, tremor, repeated episodes of consciousness loss or stroke-like episodes, parkinsonism, and autonomic/peripheral neuropathy at varying ages of onset [19]. A distinctive MRI feature is abnormally high signal intensity at the corticomedullary junction (U-fibers) on DWI and occasionally a FLAIR hyperintensity signal in the cerebellar paravermal area or middle cerebellar peduncles (Figure 3A, B) [20]. Intranuclear eosinophilic hyaline inclusions immunoreactive to anti-ubiquitin and anti-p62 in both neuronal and nonneuronal cells are a pathological hallmark [21]. A noncoding CGG repeat expansion in NOTCH2NLC was identified in 2019 as the causative mutation of NIID in Japanese patients [22,23]. Although the same expansion has been reported in several East Asian cohorts [24-26], Chen et al. [27] published a contradictory report in which no certain mutation could be found in eleven brain pathology–confirmed European NIID cases. With these conflicting findings between Asian and European populations, NIID remains a puzzle, and more investigation is certainly needed to obtain a complete picture.
- FXTAS is a late-onset neurodegenerative disorder that usually occurs after 60 years of age and has core features of intention tremor and cerebellar ataxia. Parkinsonism, executive function deficits, dementia, neuropathy, and dysautonomia are commonly associated [28,29]. All individuals with FXTAS are premutation carriers of the fragile X mental retardation 1 gene (FMR1) (55–200 CGG repeats, healthy individuals < 45 repeats), the same gene causing fragile X syndrome when in the full mutation range (> 200 CGG repeats). It primarily affects males, and females are less affected because of the presence of a normal second X chromosome. It is believed that the pathogenesis of FXTAS is related to the overexpression and “toxicity” of FMR1 mRNA [30]. The characteristic MRI features are brain atrophy and T2/FLAIR high-intensity signals in the middle cerebellar peduncles, periventricular areas, and splenium of the corpus callosum [31]. Moreover, Padilha reported FXTAS cases with abnormal high-intensity signals along the U-fibers, which have been considered unique to NIID [32]. For such similarities in clinical presentations and neuroimaging features between FXTAS and NIID, Sone et al. [19] suggested genetic analysis of the FMR1 premutation before the diagnosis of NIID.
- DRPLA is an autosomal dominant hereditary neurodegenerative disorder caused by a CAG trinucleotide repeat expansion (repeat length ≥ 48) in the atrophin-1 (ATN1) gene. An inverse correlation exists between the age at onset and the size of the CAG repeats. The cardinal features in adults are ataxia, choreoathetosis, dementia, and seizures. The onset age ranges from 0 to 72 years old, with a mean of 31 years [33]. This disease was initially mainly reported in Asian populations [34], but wider genetic testing has gradually increased the frequency of diagnosis in other ethnic groups [35]. MRI findings are characterized by progressive brainstem and cerebellum atrophy and are sometimes associated with high signal intensity in the brainstem, bilateral thalamus, and cerebral white matter in adult-onset or long disease duration patients (Figure 3C) [36,37].
- Mitochondria are unique in that they are under dual control of their own and the nuclear genome [38]. Because the main role of mitochondria is to provide an energy source for the cell, the most affected tissues are those that require the most energy, including the CNS. Owing to the widespread distribution and versatile function, the clinical spectrum can be extremely variable and have multisystem involvement. Screening for mtDNA is beyond the reach of conventional NGS. Here, two mitochondrial diseases are briefly introduced to remind clinicians of these frequently overlooked diseases after gaining a negative NGS result.
- - Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS): m.3243A>G is the most common cause of MELAS. Patients often have short stature and neurological symptoms, such as seizures, recurrent migraines, muscle weakness or exercise intolerance, and sensorineural hearing loss. Lactic acidosis and stroke-like episodes with hemiparesis and/or cortical blindness are hallmarks. Most stroke-like events begin before 40 years of age [39].
- - Myoclonic epilepsy with ragged red fibers(MERRF): m.8344A>G is the most common cause of MERRF. MERRF is clinically characterized by predominant progressive myoclonic epilepsy, and the myoclonus is often photosensitive and aggravated by action and stimuli. In addition to seizures, patients with MERRF frequently present cerebellar ataxia, mental deterioration, muscle weakness, and sensorineural hearing loss. Cardiomyopathy and arrhythmia can be seen [40].
- The “red flag” of mitochondrial disease is unexplained multisystem involvement, often including short stature, sensorineural hearing loss, migraine, cardiomyopathy, kidney problems, neuropathy, myopathy with exercise intolerance, diabetes and other endocrine disorders, and gastrointestinal dysmotility [41-43]. The most common specific MRI findings are symmetrical signal abnormalities (high T2/FLAIR and low T1 signal) of the deep gray matter, such as the basal ganglia, thalamus and brainstem [44], although these signals vary widely among different diseases. Bilateral symmetric, confluent, or multifocal white matter abnormalities can sometimes be identified. Analyzing blood lactate and pyruvate levels is the first step of the biochemical approach.
- Step 5: Genetic assay with broad-spectrum NGS
- If the initial screening of repeat expansion diseases/mtDNA fails to disclose the diagnosis, it might be appropriate to proceed to more extensive molecular genetic testing. These tools include a combination of gene-targeted testing (single-gene testing or multigene panel) and comprehensive genomic testing (whole-exome sequencing [WES], whole-genome sequencing [WGS], or chromosomal microarray analysis). When the phenotypic and neuroimaging findings suggest a characteristic diagnosis, e.g., leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL), Wilson’s disease (WD) or Alexander disease (AxD), gene-targeted testing, such as single-gene testing or a leukoencephalopathy multigene panel, can be applied. If no pathogenic variant is identified, we can proceed to gene-targeted deletion/duplication analysis, such as multiplex ligation-dependent probe amplification (MLPA). However, if the phenotype or the neuroimaging is indistinguishable from that in other GLEs, we recommend a more comprehensive NGS method with multigene panels, WES, or even WGS. As mentioned before, GLEs are a group of phenotypically and genetically heterogeneous diseases, and the number of associated genes continues to increase rapidly (Figure 1). Traditional single-gene analysis is not applicable currently due to its time-consuming nature and low yield rate. Thus, it is foreseeable that the priority order of genetic tests will change in many cases as the diagnostic success rate and cost-effectiveness of NGS continue to improve. At that time, the diagnostic algorithm could be revised again.
- Several researchers have applied multigene panels or WES to evaluate patients with leukoencephalopathy. Although the recruited patients have mainly been children, the WES experience from Vanderver et al. [45] showed an overall clinical diagnostic yield of 42% in individuals with unknown white matter abnormalities; the rate even reached 75% in a large cohort study performed in Iran [46]. After a literature review, the results of NGS studies on adult-onset GLEs are summarized as follows.
- Ayrignac et al. [47], in France (2015), classified patients into three groups according to MRI: vascular, cavitary, and others. Overall, 32 of 55 (58%) patients in the vascular group were diagnosed with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) (NOTCH3); 13 of 17 (76%) patients in the cavitary group had a diagnosis of vanishing white matter disease (VWM) (EIF2B-related disorder).
- Kunii et al. [48], in Japan (2018), identified pathogenic mutations in 8 of 60 adult patients. Five mutations were in NOTCH3, and the other three were in EIF2B2, CSF1R, and POLR3A.
- Lynch in the United Kingdom conducted two studies.
- - One hundred patients were evaluated by focused exome sequencing, and 26 cases (26%) with pathogenic or likely pathogenic variants were detected. The most frequently mutated genes were NOTCH3, EIF2B5, AARS2 and CSF1R [1].
- - Using the same method as in 2017, pathogenic or likely pathogenic variants were detected in 54 of 116 (47%) patients. The most common diagnosis was small vessel disease, followed by CSF1R mutations, mitochondrial diseases, and ALD [7].
- From the experience of these studies, it is evident that NGS is a powerful diagnostic tool for unidentified GLEs. Furthermore, CADASIL is the most frequent disease found in these large cohort studies; thus, it is crucial that clinicians remain vigilant for any hint of clinical manifestations and neuroimaging characteristics, i.e., vascular-type leukoencephalopathy. A previous study also showed that CADASIL is an important risk factor for small vessel ischemic stroke or ischemic leukoencephalopathy in Taiwan [49]. Incorporation of population-specific genetic information is rational to guide efficient molecular testing and facilitate genotype-phenotype correlation.
- However, even WES or WGS has limitations. Aside from their cost, some genetic variants, such as copy number variants (including deletion/duplication), pathogenic noncoding region (intronic) mutations, and mtDNA mutations, cannot be detected by most established NGS tools [50]. To overcome these limitations, some long-read sequencing platforms, such as Oxford Nanopore Technology and Pacific Biosciences, are being developed to enhance the diagnosis of certain repeat expansion diseases [51]. Before the development of a stable and convincing sequencing method, jumping to the conclusion that no mutation could be detected would be too hasty.
- Step 6: Big data analysis—application of artificial intelligence (AI)
- Even after using a previously described approach, a proportion of patients fail to obtain a specific diagnosis. Although knowledge of GLE is expanding and growing rapidly from worldwide experience, there are always exceptional manifestations beyond our imagination. The list of diseases related to changes in white matter is long and continues to grow; thus, it is nearly impossible for clinicians to screen such large-scale and heterogeneous data without assistance. In the current era of ever-changing technologies, we believe that AI can assist in this arduous and exhausting work. The applications of AI in medicine debuted in 2018 and have achieved astonishing success in the radiological and pathological fields [52,53]. To generate a reliable AI-based diagnostic algorithm, the top priority should be to input correct and explicit data. As clinicians, our goal is to identify the features of movement disorders and annotate them with appropriate phenomenology. In addition, detailed clinical, imaging, and biochemical information should be collected and formed into a structured input. Before the full integration of AI into day-to-day medical practice, a proper understanding and sound training of movement disorders remain the foundation to prevent future technology from going astray.
HOW TO APPROACH A PATIENT WITH MOVEMENT DISORDER AND LEUKOENCEPHALOPATHY?
NIID
Fragile X-associated tremor/ataxia syndrome (FXTAS)
Dentato-rubral-pallido-luysian atrophy (DRPLA)
MtDNA mutation diseases
Experience from prior studies: common diseases in large cohorts
- In this section, GLEs with each type of movement disorder are listed at the beginning of the corresponding subsection, followed by a review of clinical features and MRI findings in a selection of diseases.
- Parkinsonism
- - CADASIL
- - Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL)
- - CSF1R-related leukoencephalopathy
- - Cerebrotendinous xanthomatosis (CTX)
- - FXTAS
- - WD [54]
- - Neurodegeneration with brain iron accumulation (NBIA)
- - AxD [55]
- - GM1 [56]
- - Adult polyglucosan body disease (APBD)
- - Phenylketonuria (PKU) [57]
- CADASIL is an autosomal dominantly inherited small-vessel disease caused by mutations in the NOTCH3 gene on chromosome 19 [58]. It mainly affects arteries distributed in the basal ganglia and periventricular white matter, causing subcortical lacunar infarcts and the key imaging feature of symmetric confluent white matter changes in the anterior temporal pole, the external capsule, and the centrum semiovale (Figure 3D, E). Cerebral microbleeds are quite common and can be seen in one-third of individuals, mostly in the thalamus, basal ganglia, and brainstem [58-60]. In one study of then p.R544C NOTCH3 mutation in Taiwan, 5% of nonlobar intracerebral hemorrhage cases were found to be CADASIL [61].
- The most common clinical manifestations of CADASIL are migraine with aura, recurrent ischemic events (transient ischemic attacks or strokes), psychiatric symptoms, and a subcortical type of dementia, with wide variation in the age of onset [60]. Movement disorders are unusual presentations, but parkinsonism is reported most frequently among them [62,63]. Ragno et al. [64] reported five patients carrying the R1006C mutation affected by parkinsonism and demonstrated that it is a late, but not rare, feature of CADASIL.
- CARASIL is a very rare cerebral small vessel disease caused by biallelic mutations of the high-temperature requirement protease A1 (HTRA1) gene on chromosome 10q25, with most cases reported in Japan and China [65]. Except for the incidence, the clinical pictures and white matter changes of CARASIL are similar to those of CADASIL (anterior temporal, external capsules, brainstem, and cerebellum), but cognitive decline begins earlier, and the arc-shaped hyperintense lesion from the pons to the middle cerebellar peduncles (arc sign) is a characteristic finding of advanced stage [66]. In addition, gait disturbances, spondylosis, and alopecia are characteristic features. The onset of symptoms is around the age of 30.
- From the autopsy of a Belgian family with adult-onset leukoencephalopathy in 1936, CSF1R-related leukoencephalopathy was derived from both pigmentary orthochromatic leukodystrophy (POLD) and hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS) [67,68]. It is an adult-onset autosomal dominant white matter disease caused by a gene defect in CSF1R, which encodes the intracellular tyrosine kinase domain of microglia colony-stimulating factor 1 receptor. Lynch reported that CSF1R mutations account for 10% of idiopathic adult-onset leukodystrophies and implied that it is the most common adult-onset leukodystrophy [69]. The clinical phenotypes are broad but can be mainly characterized into two major categories: neuropsychiatric and motor symptoms [70]. The former category includes progressive cognitive decline, depression, apathy, anxiety, irritability, indifference, and other behavioral or personality changes. The latter includes parkinsonian symptoms, pyramidal signs (e.g., hyperreflexia, spasticity), bulbar signs (e.g., dysarthria, dysphagia), stereotypy, and frontal-type ataxia. The mean age at onset is 43 years old, with women tending to have an earlier onset than men [71,72].
- The radiological hallmarks are bilateral, symmetrical or asymmetrical, patchy or confluent T2/FLAIR hyperintensities in the periventricular, deep, and subcortical white matter of the frontal and parietal lobes. Persistent small diffusion-restricted lesions in the deep white matter (best seen on DWI series) and thinning of the corpus callosum (best seen on sagittal views) are characteristic (Figure 3F). CT imaging is applied to observe another distinctive picture, consisting of deep punctate calcifications, especially in the frontal white matter adjacent to the anterior horns of the lateral ventricles and the parietal subcortical white matter (Figure 3G) [73]. A ‘‘stepping stone” appearance can be seen on sagittal CT, while calcifications are distributed in the bifrontal pericallosal regions [70].
- CTX is a rare autosomal recessive disorder caused by mutations in the cytochrome P450 CYP27A1 gene located on chromosome 2q. CYP27A1 is a mitochondrial enzyme responsible for catalyzing multiple hydroxylation reactions in cholesterol metabolism and bile acid synthesis. Loss-of-function mutation of CYP27A1 results in a defective sterol 27-hydroxylase enzyme, abnormally high levels of cholestanol in the plasma, and accumulation of cholestanol and cholesterol throughout the body, especially in the brain (primarily white matter), lens, and tendons [74]. The clinical symptoms are chronic diarrhea, bilateral cataracts, tendon xanthomas, osteoporosis, and broad neurologic dysfunction, such as neuropsychiatric symptoms (behavioral changes, hallucinations, depression, etc.), cognitive impairment, pyramidal signs (spasticity, hyperreflexia, extensor plantar response), cerebellar signs (ataxia, dysarthria, nystagmus), peripheral neuropathy, and seizures [74]. Presentations of movement disorders are rarer, among which parkinsonism is the most frequently reported, followed by dystonia, myoclonus and postural tremor [75]. The specific MRI features are cerebral/cerebellar atrophy, extensive white matter lesions of the spinal cord, and symmetric low signal intensity in the dentate nuclei with high signal intensity within the surrounding white matter on T2WI (Figure 3H) [74,76].
- APBD, also called glycogen storage disease type IV (GSD IV), is a rare autosomal recessive GLE caused by a deficiency in glycogen branching enzyme-1 (GBE-1). Onset occurs in the fifth to seventh decades, with hallmark symptoms of neurogenic bladder, spastic paraparesis, vibration loss, and peripheral neuropathy [77]. Stroke-like events and movement disorders with parkinsonism and tremor have been reported [78-80]. The neuroimaging features are hyperintense white matter abnormalities in the periventricular regions (predominantly in the occipital lobe), the posterior limb of the internal capsule, the external capsule, the cerebellar hemisphere, the pyramidal tracts, and the medial lemniscus of the pons and medulla; the U-fibers and corpus callosum are spared. Atrophy of the medulla and cervical spine is another key universal finding [77]. This picture should be differentiated from the “tadpole-like atrophy” in Alexander disease.
- Ataxia
- - ALD
- - CSF1R-related leukoencephalopathy
- - CTX
- - LBSL
- - DRPLA
- - AxD
- - Niemann-Pick disease type C (NPC)
- - WD
- - Adult onset autosomal dominant leukodystrophy (ADLD)
- - AARS2
- - VWM
- - MLD
- - APBD
- - Krabbe disease (KD)
- - Sialidosis type I [81]
- - GM1 [56]
- - CLCN2
- - Leukoencephalopathy with calcifications and cysts (LCC)
- ALD is the most common peroxisomal metabolic disorder caused by mutations in the ATP-binding cassette subfamily D member 1 (ABCD1) gene. ABCD1 is located on the X chromosome and encodes adrenoleukodystrophy protein (ALDP), which is associated with VLCFA beta oxidation in peroxisomes. The accumulation of VLCFAs is distributed in different tissues, including the brain white matter, spinal cord, and adrenal cortex. ALD can present at different ages with a broad spectrum of phenotypic expressions due to the variable penetrance. The main phenotypes are as follows [4,82].
- - Addison’s only: 80% in childhood, 90% males and < 2% females [4].
- - Cerebral ALD: 50% in males, mostly boys between the ages of 3 and 10 years old; can be adult onset although rare. Adult men who develop the cerebral form without preceding adrenomyeloneuropathy (AMN) usually present with dementia and behavioral changes, followed by a progression course similar to that of boys [4]. White matter lesions on MRI typically begin in the splenium of the corpus callosum and then extend into the posterior limbs of the internal capsule and the periventricular and occipital white matter. Gadolinium enhancement can be seen.
- - AMN type: The second most common phenotype of ALD (40%–46%) but the most common phenotype in adults. It begins in approximately the third decade of life. It mainly involves the spinal cord and peripheral nerves and is characterized by slowly progressive spastic paraplegia and sphincter dysfunctions, often accompanied by impaired adrenocortical function. From the experience of previous reports, clinicians should bear this subtype of ALD in mind when they encounter a hereditary spastic paraplegia patient, regardless of inheritance type [83,84]. Approximately 20% to 40% of men will also present with cerebral involvement over the disease course. Approximately 50% of female carriers will develop a milder form of AMN at a later age [4]. MRI of the brain might be normal, while MRI of the spinal cord reveals diffuse atrophy.
- - Cerebello-brainstem dominant form: Rare, 1%–2% of ALD cases, mainly involving the cerebellum and brainstem, also called the “olivo-ponto-cerebellar form” or “spinocerebellar variant” (Figure 3I) [85].
- Elevated plasma VLCFA levels (C24–C26) are highly suggestive of ALD, with C26:0 and the ratios of C24/C22 and C26/ C22 as the most effective diagnostic items [82]. However, 10%–15% of female carriers have normal plasma VLCFA levels. Consequently, ABCD1 mutation screening is considered the best confirmatory test in women suspected of ALD [86].
- Mitochondrial aminoacyl-tRNA synthetases (mt-ARSs) are a group of nuclear‐encoded (ARS2 genes) enzymes catalyzing the attachment of amino acids to their cognate tRNAs, ensuring the accuracy of the mitochondrial translation process [87]. mt-ARS mutations have been linked to several neurological and neuromuscular disorders. Here, we introduce the two mutations most commonly associated with GLE: alanyl-tRNA synthetase 2 (AARS2) and aspartyl-tRNA synthetase 2 (DARS2) mutations. AARS2 leukoencephalopathy is introduced in the “Tremor” section.
- LBSL is a rare GLE caused by autosomal recessive mutations in the mitochondrial DARS2 gene [88]. The most frequent mutation variant is a splicing site mutation in intron 2 of the DARS2 gene. This region is poorly identified by NGS, and single gene-targeted testing is recommended. The onset age varies from infancy to adulthood. The main clinical features are slowly progressive pyramidal, cerebellar, and dorsal column dysfunction involving the legs more than the arms. Patients often present with spastic paraparesis and attenuated vibration sensation in the lower limbs. The unique MRI shows signal abnormalities in specific tracts in the brainstem, including the medullary pyramids, cerebellar peduncles, intraparenchymal trajectories of the trigeminal nerves, medial lemniscus, dorsal columns and lateral corticospinal tracts of the spinal cord (Figure 3J-L) [89,90]. Magnetic resonance spectroscopy shows a lactate peak. The MRI pattern is highly specific and diagnostic, but the severity of MRI changes does not correlate with the severity of disease [91].
- AxD is caused by mutations in the glial fibrillary acidic protein (GFAP) gene [92]. Although nearly all published mutations are heterozygous, the first recessive-like inheritance adult-onset AxD carrying a homozygous c.197G > A (p.R66Q) mutation was reported in a consanguineous family in Taiwan [93]. Based on the age of onset, patients are divided into three clinical subtypes: infantile (0–2 years old), juvenile (2–12 years old), and adult (> 12 years old) [94]. Typical infantile AxD is characterized by megalencephaly, developmental delay, psychomotor regression, seizure, and quadriparesis with a rapid and lethal course. Imaging of infantile AxD shows diffuse leukoencephalopathy with frontal predominance. Adult-onset AxD presents quite different clinical manifestations from those of the infantile type: bulbar symptoms, palatal myoclonus, cerebellar ataxia, spastic paraparesis, autonomic dysfunction, and sleep abnormality [95]. Distinctive atrophy of the medulla oblongata and upper cervical spinal cord with an intact pontine base (“tadpole-like atrophy”) are the characteristic imaging appearances (Figure 3M), whereas the periventricular white matter is less involved than with the infantile type [96]. Bearing in mind the characteristic clinical presentations and imaging features will assist in the diagnosis of this rare disease.
- MLD is a lysosomal storage disease caused by mutations in the ARSA or PSAP genes, resulting in deficiency of the lysosomal enzyme arylsulfatase A (ARSA) and sphingolipid activator protein B (SapB). The accumulation of sulfatides and sphingolipids in myelin leads to myelin sheath damage and destruction of nerve fibers both centrally and peripherally. The disease has an infantile form, a juvenile form, and a rarer adult form (> 16 years old). Adult MLD, the less severe subtype, presents a wide range of symptoms, including psychoses, cognitive and behavioral changes, spastic paraparesis, cerebellar ataxia, dystonia, optic atrophy, polyneuropathy, and seizures. In severe cases, MRI shows a typical appearance of “tigroid pattern” on the axial plane or “leopard pattern” on the sagittal plane, representing damage to projection fibers with sparing of the subcortical U-fibers and myelin in the perivenular areas. Diagnosis depends on genetic analysis, brain imaging, and biochemical tests of ARSA enzymatic activity in skin fibroblasts or leukocytes [97].
- VWM is caused by autosomal recessive mutations in the eukaryotic translational initiation factor 2B (EIF2B) gene. Although the onset typically occurs during childhood, 15% of cases begin in adulthood, with the oldest reported onset age being 55 years old [98,99]. Classical childhood-onset VWM mainly manifests as progressive cerebellar ataxia, with less spasticity and mental decline [100]. The adult-onset case presents a milder and more insidious clinical course with a broad range of phenotypical variabilities, including migraine, spasticity, cerebellar ataxia, behavioral changes, dementia, and seizures [101]. Women might suffer from premature ovarian insufficiency. The disease course is typically slowly progressive with episodes of deterioration provoked by stressful events (e.g., fever, infections, mild head trauma, or even fright). The neuroradiological findings in VWM are usually diagnostic, with diffuse and symmetric white matter hyperintensities in all or almost all cerebral white matter on T2WI, relatively sparing the U-fibers, and progressive rarefaction and cystic degeneration of the affected white matter [100].
- KD is an autosomal recessive lysosomal storage disorder caused by deficiency of the lysosomal enzyme galactosylceramidase (GALC). GALC deficiency leads to the storage of galactosylceramide, the formation of multinucleated macrophages (globoid cells), and the accumulation of the toxic metabolite psychosine. Accumulation of psychosine leads to widespread destruction of oligodendrocytes. Most cases have an infantile onset, but late-onset KD has been reported as well, with the oldest known age of onset being 72 years old [102]. The manifestations of late-onset KD are more variable, with spastic paraparesis the most common symptom [102]. Other features include visual loss, cognitive function impairment, ataxia, seizure and peripheral neuropathy. Brain MRI shows T2/FLAIR hyperintensities in the corticospinal tracts, occipital optic radiations, and splenium of the corpus callosum [102].
- ADLD is a rare autosomal dominant demyelinating disorder caused by duplications of the lamin B1 (LMNB1) gene. Because copy number variants are not reliably detected by NGS, specific testing for LMNB1 duplications should be arranged if such mutations are suspected. The onset typically occurs between 40 and 50 years of age. The clinical presentations are initial autonomic symptoms (urinary incontinence, constipation, orthostatic hypotension, and erectile dysfunction) followed by lower-limb-predominant pyramidal and cerebellar signs, causing spasticity and ataxia. Extrapyramidal system involvement has also been reported in some cases and is mostly manifested as tremor [103,104]. MRI shows diffuse and symmetrical supra- and infratentorial white matter changes in the motor cortex, extending downward through the pyramidal tracts, the cerebral peduncles, the pyramids of the medulla oblongata, and the cerebellar peduncles. The affected cerebral white matter usually appears in the frontal lobe first, followed by the parietal, occipital, and temporal lobes. The periventricular white matter adjacent to the lateral ventricle is typically spared or less affected than the more peripheral white matter. Spinal cord atrophy is observed and seems to correlate with early autonomic symptoms and spastic paraplegia [103].
- CLCN2-related leukoencephalopathy is a rare autosomal recessive disorder caused by mutations in the CLCN2 gene on chromosome 3q. In adults, cerebellar ataxia is the most common presentation, and other features include intention tremor (commonly in the hands), dysarthria, headache, retinopathy and male infertility. The characteristic MRI findings are confluent symmetrical white matter abnormalities involving the posterior limbs of the internal capsules, the cerebral peduncles, the dentate nucleus, and the middle cerebellar peduncles, as well as more extensive involvement of the cerebral and cerebellar white matter [105].
- NPC is an autosomal recessive lysosomal lipid storage disorder caused by mutation of either the NPC1 (95%) gene or the NPC2 gene. Although most classical forms present at between 6 and 15 years of age, adult-onset cases have been documented at an escalating frequency [106]. The clinical spectrum is also extremely heterogeneous, ranging from a perinatal rapidly fatal systemic disorder to an adult-onset chronic neurodegenerative form. The adult form mainly manifests as cerebellar ataxia, dysarthria, cognitive decline, psychiatric symptoms (e.g., schizophrenia-like psychosis), vertical supranuclear gaze palsy, seizure, and laughter-induced cataplexy. Other movement disorders, such as parkinsonism, dystonia, myoclonus, and chorea, have been reported [107]. The MRI of NPC patients can be normal or show mild atrophy in the cortex, subcortical tissue, cerebellum, and corpus callosum. Subtle parieto-occipital white matter changes are common in the early stage, and further changes can extend to the periventricular and frontal regions (Figure 3N) [108].
- Dystonia
- - WD
- - CSF1R-related leukoencephalopathy
- - CADASIL
- - NBIA
- - NPC
- - VWM
- - AARS2 [109]
- - GM1/GM2 [56]
- - LCC
- - MLD
- - CLN6 (personal observation)
- Although WD was traditionally considered a rare autosomal recessive disorder caused by pathogenic variants in the coppertransporting ATP7B gene, recent studies have found that the carrier frequency and clinical prevalence are higher than previously thought [110,111]. The ATP7B protein is involved in copper homeostasis. Dysfunction of this protein results in impaired biliary excretion of copper and abnormal accumulation of copper in the liver and extrahepatic tissues. The onset age ranges from 2 to 74 years old, with a broad spectrum of clinical manifestations that consist mainly of hepatic and neuropsychiatric symptoms [112]. Neurologically, WD is a master of disguise and is capable of displaying multiple movement disorders, mostly dystonia, tremor and parkinsonism [54], but even chorea or myoclonus has been reported [113]. Although Hitoshi proposed a characteristic MRI picture, i.e., “face of the giant panda”, in the midbrain [114], this picture is not seen very frequently; in fact, the most common radiological abnormalities are T2 hyperintensities in the putamen (particularly on the lateral side), caudate nucleus, globus pallidus, thalamus, midbrain, and pons (Figure 3O) [115,116]. In more advanced cases, hyperintense areas in the midbrain, cerebellum, and subcortical white matter can be seen on T1WI.
- NBIA is a heterogeneous group of inherited disorders with abnormal brain iron accumulation [117]. The deposits of excess iron are distributed mainly in the globus pallidus and substantia nigra (SN). Progressive extrapyramidal movement disorders (mainly dystonia and parkinsonism), dysarthria, spasticity, neuropsychiatric abnormalities, and eye problems (optic atrophy or retinal degeneration) are the characteristic clinical manifestations [118]. Pantothenate kinase-associated neurodegeneration (PKAN) is the most common form and accounts for more than 50% of NBIA cases [119]. Apart from the eye-of-the-tiger sign (a round hyperintense center with surrounding hypointensity in the globus pallidus on T2WI), the hallmark imaging feature of PKAN, periventricular and subcortical white matter hyperintensity on T2WI, can be seen in phospholipase-associated neurodegeneration (PLAN), fatty acid hydroxylase-associated neurodegeneration (FAHN), aceruloplasminemia, and Woodhouse-Sakati syndrome. A hyperintense halo surrounding a central band of hypointensity in the SN on axial T1WI is characteristic of beta-propeller protein-associated neurodegeneration (BPAN) [120-122].
- Tremor
- The GLEs with tremor are listed below; most of these diseases, excluding AARS2 leukoencephalopathy, have been introduced in other sections.
- - WD
- - FXTAS
- - NIID
- - CSF1R-related leukoencephalopathy
- - AARS2 leukoencephalopathy
- - CLCN2-related leukoencephalopathy [123]
- - VWM [124]
- - ADLD [104]
- - PKU [125]
- AARS2 leukoencephalopathy is caused by autosomal recessive mutations in the AARS2 gene. Two very different phenotypes have been reported: late-onset ovarian failure with leukodystrophy and early-onset cardiomyopathy [126]. The average age of adult-onset leukoencephalopathy is 27.3 years old. The most common clinical manifestations are psychiatric symptoms, cognitive decline, pyramidal signs, parkinsonism, ataxia and seizure. Ovarian failure is a distinguishing feature in female patients [127]. Surprisingly, many clinical phenotypes, imaging features, and even pathological hallmarks of AARS2 leukoencephalopathy overlap with those of CSF1R-related leukoencephalopathy [128]. Lynch even suggested screening for AARS2 mutations in all patients with clinical or radiological features resembling those of CSF1R-related leukoencephalopathy [129].
- Chorea
- The GLEs with chorea are listed below; most of these diseases have been introduced in other sections.
- - DRPLA [33]
- - WD [130]
- - NBIA [131]
- - MLD [132]
- - AARS2 leukoencephalopathy [109]
- Myoclonus
- The GLEs with myoclonus are listed below; most of these diseases have been introduced in other sections.
- - DRPLA [33]
- - MERRF [40]
- - WD [113]
- - CSF1R-related leukoencephalopathy [69]
- - NBIA [118]
- - AxD (palatal myoclonus) [95]
- - VWM [133]
- - Sialidosis type I [134]
- Stereotype
- The GLEs with stereotype are listed below; most of these diseases, excluding leukoencephalopathy with calcifications and cysts, have been introduced in other sections.
- - LCC
- - CSF1R-related leukoencephalopathy [135]
- - WD [136]
- - MLD [137]
- - Neuroferritinopathy (NBIA) [138]
- - AARS2 [126]
- LCC is a rare autosomal recessive disease caused by biallelic mutations in the SNORD118 gene [139]. It is also known as Labrune syndrome after the author who first reported it in 1996 [140]. The diagnosis depends on a characteristic radiological triad: diffuse and asymmetric brain calcifications, formation of parenchymal cysts, and abnormal white matter density/signal. The age of onset can range from early infancy to adolescence or even adulthood, and the neurological manifestations are highly variable, including developmental delay, seizures, headaches, cognitive decline, visual changes, speech disturbances, ataxia, pyramidal signs, extrapyramidal symptoms (stereotypy and dystonia), and ischemic/hemorrhagic strokes [139,141].
THE INTRODUCTION OF EACH DISEASE (EXCLUDING REPEAT EXPANSION DISORDERS) ASSOCIATED WITH DIFFERENT MOVEMENT DISORDER ENTITIES
CADASIL
CARASIL
CSF1R-related leukoencephalopathy
CTX
APBD
X-linked ALD
LBSL
AxD
MLD
VWM
KD
ADLD
CLCN2-related leukoencephalopathy
NPC
WD
NBIA
AARS2 leukoencephalopathy
LCC
- There is no cure for most GLEs, and only supportive treatment is available. Symptomatic therapies include antidepressants for depression, muscle relaxants for spasticity, antiepileptic drugs for epilepsy, Botulinum toxin injection for dystonia, and physical/occupational therapy to minimize muscle tone deterioration and daily activity dependence. Although treatments to prolong remission are lacking in most conditions, early detection of certain diseases is crucial for the provision of disease-specific therapy, listed in Table 2.
TREATMENT STRATEGY
- GLEs are a group of disorders with heterogeneous causes, clinical presentations, and radiological features. These conditions are often difficult to diagnose, and the mix of movement disorders complicates the situation even further. Here, we have attempted to provide an algorithm for practicing neurologists to approach GLEs (Figure 4), but it is imperative to bear in mind that some patients still cannot be definitively diagnosed even with this approach. The heterogeneity of disease manifestations and genetic penetrance always surpass our imagination. In this era of rapidly evolving technology, the processing speed of big data analytics continues to improve, and its application in medical use is anticipated. Clinicians should take full advantage of these advanced tools when facing diagnostic challenges. To achieve a reliable interpretation, we are responsible for providing detailed and accurate clinical information, radiological manifestations, laboratory findings, or even results from genetic tests. With the assistance of AI technology, we expect that the investigation of GLEs with movement disorders will become more efficient in the foreseeable future.
CONCLUSION
-
Conflicts of Interest
The authors have no financial conflicts of interest.
-
Funding Statement
None
-
Author contributions
Conceptualization: Mu-Hui Fu, Yung-Yee Chang. Data curation: Mu-Hui Fu, Yung-Yee Chang. Investigation: Mu-Hui Fu, Yung-Yee Chang. Methodology: Mu-Hui Fu, Yung-Yee Chang. Project administration: Mu-Hui Fu. Resources: Mu-Hui Fu, Yung-Yee Chang. Software: Mu-Hui Fu. Supervision: Yung-Yee Chang. Validation: Mu-Hui Fu, Yung-Yee Chang. Visualization: Mu-Hui Fu, Yung-Yee Chang. Writing—original draft: Mu-Hui Fu. Writing—review & editing: Mu-Hui Fu.
Notes
- We would like to express our gratitude to Dr. Chin-Hsien Lin, Professor of Department of Neurology in National Taiwan University Hospital for courtesy of providing the imaging of a Niemann Pick disease type C patient.
Acknowledgments

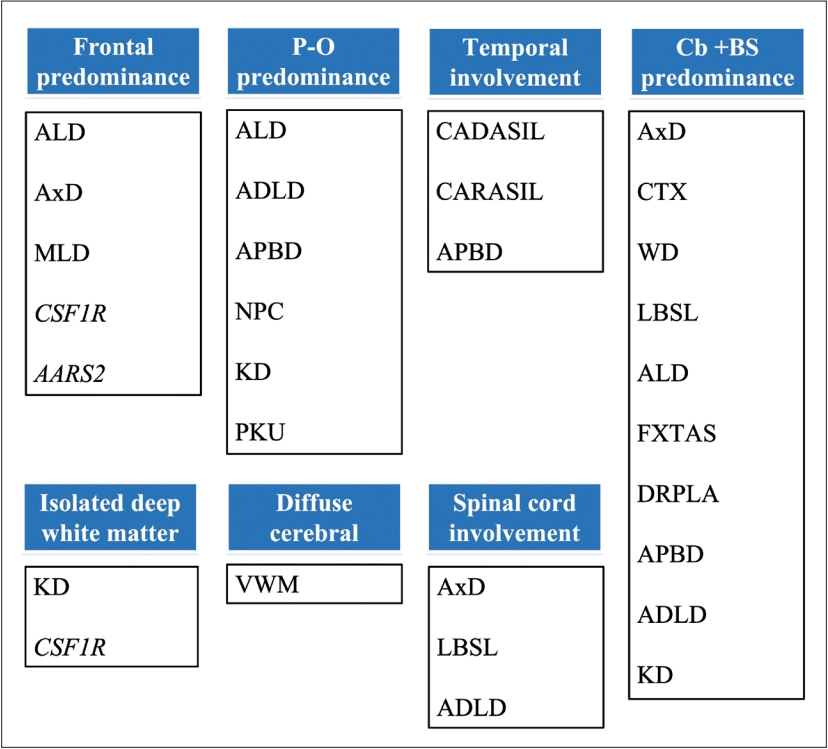
 indicates the presence of characteristic findings or a positive result.
indicates the presence of characteristic findings or a positive result.  indicates the absence of characteristic findings or a negative result. Hx, history; C/T, chemotherapy; R/T, radiotherapy; BP, blood pressure; MRI, magnetic resonance imaging; CT, computed tomography; C+-, with/without enhancement; Cu, copper; CSF, cerebrospinal fluid; NCV, nerve conduction velocity; VLCFA, very-long-chain fatty acid; WCE, white cell enzymes; mito, mitochondrial; WES, whole-exome sequencing; WGS, whole-genome sequencing; GLE, genetic leukoencephalopathy; AxD, Alexander disease; CADASIL, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CSF1R, CSF1R-related leukoencephalopathy; CTX, cerebrotendinous xanthomatosis; DRPLA, dentatorubral-pallidoluysian atrophy; FXTAS, fragile X-associated tremor/ataxia syndrome; LBSL, leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation; NBIA, neurodegeneration with brain iron accumulation; NIID, neuronal intranuclear inclusion disease; WD, Wilson’s disease; S/S, symptoms/signs.
indicates the absence of characteristic findings or a negative result. Hx, history; C/T, chemotherapy; R/T, radiotherapy; BP, blood pressure; MRI, magnetic resonance imaging; CT, computed tomography; C+-, with/without enhancement; Cu, copper; CSF, cerebrospinal fluid; NCV, nerve conduction velocity; VLCFA, very-long-chain fatty acid; WCE, white cell enzymes; mito, mitochondrial; WES, whole-exome sequencing; WGS, whole-genome sequencing; GLE, genetic leukoencephalopathy; AxD, Alexander disease; CADASIL, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CSF1R, CSF1R-related leukoencephalopathy; CTX, cerebrotendinous xanthomatosis; DRPLA, dentatorubral-pallidoluysian atrophy; FXTAS, fragile X-associated tremor/ataxia syndrome; LBSL, leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation; NBIA, neurodegeneration with brain iron accumulation; NIID, neuronal intranuclear inclusion disease; WD, Wilson’s disease; S/S, symptoms/signs.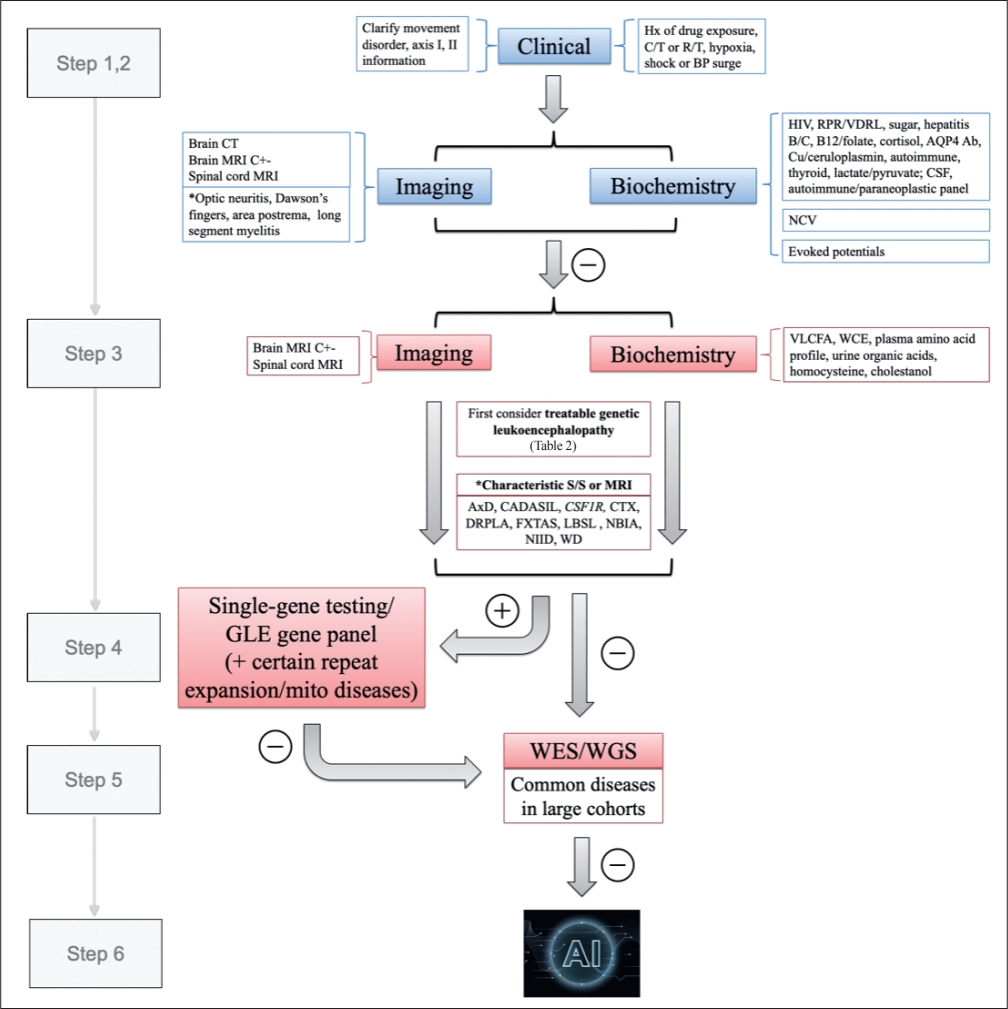
| Disease | Inheritance | Gene | MRI characteristics |
|---|---|---|---|
| AARS2 leukoencephalopathy | AR | AARS2 | Periventricular rarefaction, frontoparietal periventricular deep white matter; punctate areas of restricted diffusion (should differentiate from CSF1R-related leukoencephalopathy) |
| ADLD | AD | LMNB1 | Symmetric white matter extending from motor cortex, corticospinal tracts, downward through the posterior limb of the internal capsule, toward the medullary pyramids |
| ALD | X-linked | ABCD1 | Cerebral form: splenium of the corpus callosum, periventricular regions with P-O predominance, visual and auditory pathways |
| AMN: diffuse spinal cord atrophy, particularly T-spine; corticospinal tract involvement | |||
| APBD | AR | GBE1 | Periventricular regions (occipital predominance), posterior limb of the internal capsule, cerebellar hemisphere, medial lemniscus of the pons and medulla; atrophy of the medulla and cervical spine (should differentiate from AxD) |
| AxD | AD, AR [93] | GFAP | Atrophy of medulla oblongata and upper cervical cord (tadpole brainstem atrophy) |
| CADASIL | AD | NOTCH3 | External capsule, temporal poles, subcortical lacunes, cerebral microbleeds CADASIL-alike, arc sign suggests advanced stage |
| CARASIL | AR | HTRA1 | Bilateral, symmetrical/asymmetrical, patchy/confluent frontoparietal |
| CSF1R-related leukoencephalopathy | AD | CSF1R | periventricular white matter; frontal involvement with restricted diffusion; thinning of corpus callosum; small cysts and periventricular calcifications |
| CLCN2-related leukoencephalopathy | AR | CLCN2 | Symmetric posterior limbs of internal capsules, cerebral peduncles, and middle cerebellar peduncles |
| CTX | AR | CYP27A1 | Deep cerebellar white matter and dentate nucleus |
| DRPLA | AD | ATN1 | Progressive BS and Cb atrophy; high intensity in the BS, bilateral thalami, and cerebral white matter in the elderly |
| FXTAS | X-linked | Premutation expansion of FMR1 | Middle cerebellar peduncles, periventricular areas, splenium of the corpus callosum; abnormal high-intensity signal along the U-fibers |
| GM1 gangliosidoses | AR | GLB1 | (should differentiate from NIID) |
| KD | AR | GALC | Pallidal hypointensity and bilateral symmetrical putaminal hyperintensities (especially posterior part) |
| LBSL | AR | DARS2 | Corticospinal tracts, callosal splenium, and posterior periventricular and deep white matter (optic radiation) |
| LCC | AR | SNORD118 | Posterior limbs of the internal capsules, tracts and trajectories of the trigeminal nerves, pyramids of medulla oblongata, decussation of the medial lemniscus; dorsal columns and lateral corticospinal tracts of the medulla oblongata or spinal cord |
| MLD | AR | ARSA or PSAP | Diffuse and asymmetric brain calcifications, formation of parenchymal cysts “Tigroid” or “leopard-skin” pattern |
| MtDNA mutation diseases | Maternal inheritance | Deep gray matter and white matter abnormalities | |
| NBIA | AD, AR, X-linked | PANK2, WDR45, PLA2G6, FA2H, CP, DCAF17 | PKAN: eye-of-the-tiger sign |
| BPAN: SN halo sign in T1WI | |||
| PLAN, FAHN, aceruloplasminemia, Woodhouse-Sakati syndrome: periventricular and subcortical white matter | |||
| NIID | AD | NOTCH2NLC | DWI high-intensity signals in the corticomedullary junction along U-fibers Subtle white matter change in the P-O region |
| NPC VWM | AR AR | NPC1 or NPC2 EIF2B | Diffuse and symmetric white matter abnormalities, progressive rarefaction and cystic degeneration |
| WD | AR | ATP7B | T2 hyperintensities in putamen (particularly lateral side) and caudate nucleus; “face of the giant panda” in the midbrain |
Leukoencephalopathy with enhancement: X-ALD, AxD, Krabbe disease.
ADLD, adult onset autosomal dominant leukodystrophy; ALD, adrenoleukodystrophy; APBD, adult polyglucosan body disease; AxD, Alexander disease; CADASIL, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CARASIL, cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy; CTX, cerebrotendinous xanthomatosis; DRPLA, dentatorubral-pallidoluysian atrophy; FXTAS, fragile X-associated tremor/ataxia syndrome; KD, Krabbe disease; LBSL, leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation; LCC, leukoencephalopathy with calcifications and cysts; MLD, metachromatic leukodystrophy; MtDNA, mitochondrial DNA; NBIA, neurodegeneration with brain iron accumulation; NIID, neuronal intranuclear inclusion disease; NPC, Niemann Pick disease type C; VWM, vanishing white matter disease; WD, Wilson’s disease; BS, brainstem; Cb, cerebellum; CP, ceruloplasmin; P-O, parieto-occipital; SN, substantia nigra; T1WI, T1-weighted imaging; AMN, adrenomyeloneuropathy; PKAN, pantothenate kinase-associated neurodegeneration; BPAN, beta-propeller protein-associated neurodegeneration; PLAN, phospholipase-associated neurodegeneration; FAHN, fatty acid hydroxylase-associated neurodegeneration; DWI, diffusion-weighted imaging.
- 1. Lynch DS, Rodrigues Brandão de Paiva A, Zhang WJ, Bugiardini E, Freua F, Tavares Lucato L, et al. Clinical and genetic characterization of leukoencephalopathies in adults. Brain 2017;140:1204–1211.ArticlePubMedPMC
- 2. Vanderver A, Prust M, Tonduti D, Mochel F, Hussey HM, Helman G, et al. Case definition and classification of leukodystrophies and leukoencephalopathies. Mol Genet Metab 2015;114:494–500.ArticlePubMedPMC
- 3. Helman G, Venkateswaran S, Vanderver A. The spectrum of adult-onset heritable white-matter disorders. Handb Clin Neurol 2018;148:669–692.ArticlePubMed
- 4. Renaud DL. Adult-onset leukoencephalopathies. Continuum (Minneap Minn) 2016;22(2 Dementia):559–578.ArticlePubMed
- 5. Johns Hopkins University. OMIM(R). An Online Catalog of Human Genes and Genetic Disorders. Search: ‘leukodystrophy or leukoencephalopathy’ [Internet]. Baltimore: Johns Hopkins University; c1966- 2023 [accessed on 2022 July 21]. Available at: https://www.omim.org/search?index=entry&search=leukodystrophy+or+leukoencephalopathy&sort=score+desc%2C+prefix_sort+desc&start=1&limit=10.
- 6. Köhler W, Curiel J, Vanderver A. Adulthood leukodystrophies. Nat Rev Neurol 2018;14:94–105.ArticlePubMedPDF
- 7. Lynch DS, Wade C, Paiva ARB, John N, Kinsella JA, Merwick Á, et al. Practical approach to the diagnosis of adult-onset leukodystrophies: an updated guide in the genomic era. J Neurol Neurosurg Psychiatry 2019;90:543–554.ArticlePubMed
- 8. Williams T, Houlden H, Murphy E, John N, Fox NC, Schott JM, et al. How to diagnose difficult white matter disorders. Pract Neurol 2020;20:280–286.ArticlePubMed
- 9. van der Knaap MS, Schiffmann R, Mochel F, Wolf NI. Diagnosis, prognosis, and treatment of leukodystrophies. Lancet Neurol 2019;18:962–972.ArticlePubMed
- 10. Abdo WF, van de Warrenburg BP, Burn DJ, Quinn NP, Bloem BR. The clinical approach to movement disorders. Nat Rev Neurol 2010;6:29–37.ArticlePubMedPDF
- 11. Aerts MB, Jankovic J, van de Warrenburg BP, Bloem BR. Phenomenology, classification, and diagnostic approach to patients with movement disorders. In: Poewe W, Jankovic J, editors. Movement Disorders in Neurologic and Systemic Disease. 1st ed. Cambridge: Cambridge University Press; 2014:1–15.
- 12. Albanese A, Bhatia K, Bressman SB, Delong MR, Fahn S, Fung VS, et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord 2013;28:863–873.ArticlePubMedPMCPDF
- 13. Ahmed RM, Murphy E, Davagnanam I, Parton M, Schott JM, Mummery CJ, et al. A practical approach to diagnosing adult onset leukodystrophies. J Neurol Neurosurg Psychiatry 2014;85:770–781.ArticlePubMed
- 14. Sindhwani G, Arora M, Thakker VD, Jain A. MRI in chemotherapy induced leukoencephalopathy: report of two cases and radiologist’s perspective. J Clin Diagn Res 2017;11:TD08–TD09.ArticlePubMedPMC
- 15. Kim SM, Kim SJ, Lee HJ, Kuroda H, Palace J, Fujihara K. Differential diagnosis of neuromyelitis optica spectrum disorders. Ther Adv Neurol Disord 2017;10:265–289.ArticlePubMedPMCPDF
- 16. Moser HW, Mahmood A, Raymond GV. X-linked adrenoleukodystrophy. Nat Clin Pract Neurol 2007;3:140–151.ArticlePubMedPDF
- 17. Parikh S, Bernard G, Leventer RJ, van der Knaap MS, van Hove J, Pizzino A, et al. A clinical approach to the diagnosis of patients with leukodystrophies and genetic leukoencephelopathies. Mol Genet Metab 2015;114:501–515.ArticlePubMedPMC
- 18. Paulson H. Repeat expansion diseases. Handb Clin Neurol 2018;147:105–123.ArticlePubMedPMC
- 19. Sone J, Mori K, Inagaki T, Katsumata R, Takagi S, Yokoi S, et al. Clinicopathological features of adult-onset neuronal intranuclear inclusion disease. Brain 2016;139(Pt 12):3170–3186.ArticlePubMedPMC
- 20. Sugiyama A, Sato N, Kimura Y, Maekawa T, Enokizono M, Saito Y, et al. MR imaging features of the cerebellum in adult-onset neuronal intranuclear inclusion disease: 8 cases. AJNR Am J Neuroradiol 2017;38:2100–2104.ArticlePubMedPMC
- 21. Liu Y, Mimuro M, Yoshida M, Hashizume Y, Niwa H, Miyao S, et al. Inclusion-positive cell types in adult-onset intranuclear inclusion body disease: implications for clinical diagnosis. Acta Neuropathol 2008;116:615–623.ArticlePubMedPDF
- 22. Ishiura H, Shibata S, Yoshimura J, Suzuki Y, Qu W, Doi K, et al. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat Genet 2019;51:1222–1232.ArticlePubMedPDF
- 23. Sone J, Mitsuhashi S, Fujita A, Mizuguchi T, Hamanaka K, Mori K, et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat Genet 2019;51:1215–1221.ArticlePubMedPDF
- 24. Deng J, Gu M, Miao Y, Yao S, Zhu M, Fang P, et al. Long-read sequencing identified repeat expansions in the 5’UTR of the NOTCH2NLC gene from Chinese patients with neuronal intranuclear inclusion disease. J Med Genet 2019;56:758–764.ArticlePubMed
- 25. Sun QY, Xu Q, Tian Y, Hu ZM, Qin LX, Yang JX, et al. Expansion of GGC repeat in the human-specific NOTCH2NLC gene is associated with essential tremor. Brain 2020;143:222–233.ArticlePubMedPDF
- 26. Tian WT, Zhan FX, Liu Q, Luan XH, Zhang C, Shang L, et al. Clinicopathologic characterization and abnormal autophagy of CSF1R-related leukoencephalopathy. Transl Neurodegener 2019;8:32.ArticlePubMedPMCPDF
- 27. Chen Z, Yan Yau W, Jaunmuktane Z, Tucci A, Sivakumar P, Gagliano Taliun SA, et al. Neuronal intranuclear inclusion disease is genetically heterogeneous. Ann Clin Transl Neurol 2020;7:1716–1725.ArticlePubMedPMCPDF
- 28. Hagerman RJ, Leehey M, Heinrichs W, Tassone F, Wilson R, Hills J, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 2001;57:127–130.ArticlePubMed
- 29. Cabal-Herrera AM, Tassanakijpanich N, Salcedo-Arellano MJ, Hagerman RJ. Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS): pathophysiology and clinical implications. Int J Mol Sci 2020;21:4391.ArticlePubMedPMC
- 30. Hagerman RJ, Hagerman P. Fragile X-associated tremor/ataxia syndrome-features, mechanisms and management. Nat Rev Neurol 2016;12:403–412.ArticlePubMedPDF
- 31. Brunberg JA, Jacquemont S, Hagerman RJ, Berry-Kravis EM, Grigsby J, Leehey MA, et al. Fragile X premutation carriers: characteristic MR imaging findings of adult male patients with progressive cerebellar and cognitive dysfunction. AJNR Am J Neuroradiol 2002;23:1757–1766.PubMedPMC
- 32. Padilha IG, Nunes RH, Scortegagna FA, Pedroso JL, Marussi VH, Rodrigues Gonçalves MR, et al. MR imaging features of adult-onset neuronal intranuclear inclusion disease may be indistinguishable from Fragile X-Associated Tremor/Ataxia Syndrome. AJNR Am J Neuroradiol 2018;39:E100–E101.ArticlePubMedPMC
- 33. Carroll LS, Massey TH, Wardle M, Peall KJ. Dentatorubral-pallidoluysian atrophy: an update. Tremor Other Hyperkinet Mov (N Y) 2018;8:577.ArticlePubMedPMC
- 34. Tsuji S. Dentatorubral-pallidoluysian atrophy (DRPLA): clinical features and molecular genetics. Adv Neurol 1999;79:399–409.PubMed
- 35. Wardle M, Morris HR, Robertson NP. Clinical and genetic characteristics of non-Asian dentatorubral-pallidoluysian atrophy: a systematic review. Mov Disord 2009;24:1636–1640.ArticlePubMed
- 36. Yoon WT, Youn J, Cho JW. Is cerebral white matter involvement helpful in the diagnosis of dentatorubral-pallidoluysian atrophy? J Neurol 2012;259:1694–1697.ArticlePubMedPDF
- 37. Sugiyama A, Sato N, Nakata Y, Kimura Y, Enokizono M, Maekawa T, et al. Clinical and magnetic resonance imaging features of elderly onset dentatorubral-pallidoluysian atrophy. J Neurol 2018;265:322–329.ArticlePubMedPDF
- 38. Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet 2005;6:389–402.ArticlePubMedPMCPDF
- 39. Pia S, Lui F. Melas syndrome. [updated 2022 Jul 4]. In: StatPearls [Internet]. Treasure Island: StatPearls Publishing; c2022 [accessed on 2022 Mar 2]. Available at: https://www.ncbi.nlm.nih.gov/books/NBK532959/.
- 40. Hameed S, Tadi P. Myoclonic epilepsy and ragged red fibers. [updated 2022 Jul 4]. In: StatPearls [Internet]. Treasure Island: StatPearls Publishing; c2022 [accessed on 2022 Mar 10]. Available at: https://www.ncbi.nlm.nih.gov/books/NBK555923/.
- 41. Boggan RM, Lim A, Taylor RW, McFarland R, Pickett SJ. Resolving complexity in mitochondrial disease: toward precision medicine. Mol Genet Metab 2019;128(1-2):19–29.PubMed
- 42. Keogh MJ, Chinnery PF. How to spot mitochondrial disease in adults. Clin Med (Lond) 2013;13:87–92.ArticlePubMedPMC
- 43. Liang C, Ahmad K, Sue CM. The broadening spectrum of mitochondrial disease: shifts in the diagnostic paradigm. Biochim Biophys Acta 2014;1840:1360–1367.ArticlePubMed
- 44. Wong LJ. Mitochondrial syndromes with leukoencephalopathies. Semin Neurol 2012;32:55–61.ArticlePubMed
- 45. Vanderver A, Simons C, Helman G, Crawford J, Wolf NI, Bernard G, et al. Whole exome sequencing in patients with white matter abnormalities. Ann Neurol 2016;79:1031–1037.ArticlePubMedPMCPDF
- 46. Mahdieh N, Soveizi M, Tavasoli AR, Rabbani A, Ashrafi MR, Kohlschütter A, et al. Genetic testing of leukodystrophies unraveling extensive heterogeneity in a large cohort and report of five common diseases and 38 novel variants. Sci Rep 2021;11:3231.ArticlePubMedPMCPDF
- 47. Ayrignac X, Carra-Dalliere C, Menjot de Champfleur N, Denier C, Aubourg P, Bellesme C, et al. Adult-onset genetic leukoencephalopathies: a MRI pattern-based approach in a comprehensive study of 154 patients. Brain 2015;138(Pt 2):284–292.ArticlePubMed
- 48. Kunii M, Doi H, Ishii Y, Ohba C, Tanaka K, Tada M, et al. Genetic analysis of adult leukoencephalopathy patients using a custom-designed gene panel. Clin Genet 2018;94:232–238.ArticlePubMedPDF
- 49. Lee YC, Chung CP, Chang MH, Wang SJ, Liao YC. NOTCH3 cysteinealtering variant is an important risk factor for stroke in the Taiwanese population. Neurology 2020;94:e87–e96.ArticlePubMed
- 50. Xing Y, Dabney AR, Li X, Wang G, Gill CA, Casola C. SECNVs: a simulator of copy number variants and whole-exome sequences from reference genomes. Front Genet 2020;11:82.ArticlePubMedPMC
- 51. Chintalaphani SR, Pineda SS, Deveson IW, Kumar KR. An update on the neurological short tandem repeat expansion disorders and the emergence of long-read sequencing diagnostics. Acta Neuropathol Commun 2021;9:98.ArticlePubMedPMCPDF
- 52. Liu Y, Kohlberger T, Norouzi M, Dahl GE, Smith JL, Mohtashamian A, et al. Artificial intelligence-based breast cancer nodal metastasis detection: insights into the black box for pathologists. Arch Pathol Lab Med 2019;143:859–868.ArticlePubMedPDF
- 53. Nam JG, Park S, Hwang EJ, Lee JH, Jin KN, Lim KY, et al. Development and validation of deep learning-based automatic detection algorithm for malignant pulmonary nodules on chest radiographs. Radiology 2019;290:218–228.ArticlePubMed
- 54. Hedera P. Wilson’s disease: a master of disguise. Parkinsonism Relat Disord 2019;59:140–145.ArticlePubMed
- 55. Park J, Park ST, Kim J, Kwon KY. A case report of adult-onset Alexander disease clinically presenting as Parkinson’s disease: is the comorbidity associated with genetic susceptibility? BMC Neurol 2020;20:27.ArticlePubMedPMCPDF
- 56. Regier DS, Proia RL, D’Azzo A, Tifft CJ. The GM1 and GM2 gangliosidoses: natural history and progress toward therapy. Pediatr Endocrinol Rev 2016;13 Suppl 1(Suppl 1):663–673.PubMed
- 57. Liu Y, Dong Z, Yu S. Late-diagnosed phenylketonuria mimicking x-linked adrenoleukodystrophy with heterozygous mutations of the PAH Gene: a case report and literature review. Clin Neurol Neurosurg 2018;171:151–155.ArticlePubMed
- 58. Kalimo H, Ruchoux MM, Viitanen M, Kalaria RN. CADASIL: a common form of hereditary arteriopathy causing brain infarcts and dementia. Brain Pathol 2002;12:371–384.ArticlePubMed
- 59. Domenga V, Fardoux P, Lacombe P, Monet M, Maciazek J, Krebs LT, et al. Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev 2004;18:2730–2735.ArticlePubMedPMC
- 60. Di Donato I, Bianchi S, De Stefano N, Dichgans M, Dotti MT, Duering M, et al. Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) as a model of small vessel disease: update on clinical, diagnostic, and management aspects. BMC Med 2017;15:41.ArticlePubMedPMCPDF
- 61. Tang SC, Chen YR, Chi NF, Chen CH, Cheng YW, Hsieh FI, et al. Prevalence and clinical characteristics of stroke patients with p.R544C NOTCH3 mutation in Taiwan. Ann Clin Transl Neurol 2018;6:121–128.ArticlePubMedPMCPDF
- 62. Spagnolo F, Pinto V, Rini AM, Passarella B. Dystonia and parkinsonism as presenting CADASIL features: a case report. Neurol Sci 2021;42:4781–4783.ArticlePubMedPDF
- 63. Erro R, Lees AJ, Moccia M, Picillo M, Penco S, Mosca L, et al. Progressive parkinsonism, balance difficulties, and supranuclear gaze palsy. JAMA Neurol 2014;71:104–107.ArticlePubMed
- 64. Ragno M, Berbellini A, Cacchiò G, Manca A, Di Marzio F, Pianese L, et al. Parkinsonism is a late, not rare, feature of CADASIL: a study on Italian patients carrying the R1006C mutation. Stroke 2013;44:1147–1149.ArticlePubMed
- 65. Tikka S, Baumann M, Siitonen M, Pasanen P, Pöyhönen M, Myllykangas L, et al. CADASIL and CARASIL. Brain Pathol 2014;24:525–544.PubMedPMC
- 66. Nozaki H, Sekine Y, Fukutake T, Nishimoto Y, Shimoe Y, Shirata A, et al. Characteristic features and progression of abnormalities on MRI for CARASIL. Neurology 2015;85:459–463.ArticlePubMed
- 67. Marotti JD, Tobias S, Fratkin JD, Powers JM, Rhodes CH. Adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia: report of a family, historical perspective, and review of the literature. Acta Neuropathol 2004;107:481–488.ArticlePubMedPDF
- 68. Van Bogaert L, Nyssen R. [Late filetype of familial progressive leukodystrophy (Filetype tardif de la leukodystrophie progressive familiale)]. Rev Neurol 1936;65:21–45.
- 69. Lynch DS, Jaunmuktane Z, Sheerin UM, Phadke R, Brandner S, Milonas I, et al. Hereditary leukoencephalopathy with axonal spheroids: a spectrum of phenotypes from CNS vasculitis to parkinsonism in an adult onset leukodystrophy series. J Neurol Neurosurg Psychiatry 2016;87:512–519.ArticlePubMedPMC
- 70. Konno T, Kasanuki K, Ikeuchi T, Dickson DW, Wszolek ZK. CSF1R-related leukoencephalopathy: a major player in primary microgliopathies. Neurology 2018;91:1092–1104.ArticlePubMedPMC
- 71. Ikeuchi T, Mezaki N, Miura T. Cognitive dysfunction and symptoms of movement disorders in adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. Parkinsonism Relat Disord 2018;46 Suppl 1:S39–S41.ArticlePubMed
- 72. Lan MY, Liu JS, Chang CC, Chen YF, Su CS, Peng CH, et al. Clinicopathologic and genetic studies of 2 patients with hereditary diffuse leukoencephalopathy with axonal spheroids. Alzheimer Dis Assoc Disord 2016;30:73–76.ArticlePubMed
- 73. Codjia P, Ayrignac X, Mochel F, Mouzat K, Carra-Dalliere C, Castelnovo G, et al. Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia: an MRI study of 16 French cases. AJNR Am J Neuroradiol 2018;39:1657–1661.ArticlePubMedPMC
- 74. Salen G, Steiner RD. Epidemiology, diagnosis, and treatment of cerebrotendinous xanthomatosis (CTX). J Inherit Metab Dis 2017;40:771–781.ArticlePubMedPDF
- 75. Stelten BML, van de Warrenburg BPC, Wevers RA, Verrips A. Movement disorders in cerebrotendinous xanthomatosis. Parkinsonism Relat Disord 2019;58:12–16.ArticlePubMed
- 76. Yanagihashi M, Kano O, Terashima T, Kawase Y, Hanashiro S, Sawada M, et al. Late-onset spinal form xanthomatosis without brain lesion: a case report. BMC Neurol 2016;16:21.ArticlePubMedPMC
- 77. Mochel F, Schiffmann R, Steenweg ME, Akman HO, Wallace M, Sedel F, et al. Adult polyglucosan body disease: natural history and key magnetic resonance imaging findings. Ann Neurol 2012;72:433–441.ArticlePubMedPMC
- 78. Johal J, Castro Apolo R, Johnson MW, Persch MR, Edwards A, Varade P, et al. Adult polyglucosan body disease: an acute presentation leading to unmasking of this rare disorder. Hosp Pract (1995) 2022;50:244–250.ArticlePubMed
- 79. Robertson NP, Wharton S, Anderson J, Scolding NJ. Adult polyglucosan body disease associated with an extrapyramidal syndrome. J Neurol Neurosurg Psychiatry 1998;65:788–790.ArticlePubMedPMC
- 80. Trivedi JR, Wolfe GI, Nations SP, Burns DK, Bryan WW, Dewey RB Jr. Adult polyglucosan body disease associated with lewy bodies and tremor. Arch Neurol 2003;60:764–766.ArticlePubMed
- 81. Caciotti A, Melani F, Tonin R, Cellai L, Catarzi S, Procopio E, et al. Type I sialidosis, a normosomatic lysosomal disease, in the differential diagnosis of late-onset ataxia and myoclonus: an overview. Mol Genet Metab 2020;129:47–58.ArticlePubMed
- 82. Engelen M, Kemp S, de Visser M, van Geel BM, Wanders RJ, Aubourg P, et al. X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J Rare Dis 2012;7:51.ArticlePubMedPMC
- 83. Zhan ZX, Liao XX, Du J, Luo YY, Hu ZT, Wang JL, et al. Exome sequencing released a case of X-linked adrenoleukodystrophy mimicking recessive hereditary spastic paraplegia. Eur J Med Genet 2013;56:375–378.ArticlePubMed
- 84. Shaw-Smith CJ, Lewis SJ, Reid E. X-linked adrenoleukodystrophy presenting as autosomal dominant pure hereditary spastic paraparesis. J Neurol Neurosurg Psychiatry 2004;75:686–688.ArticlePubMedPMC
- 85. Ogaki K, Koga S, Aoki N, Lin W, Suzuki K, Ross OA, et al. Adult-onset cerebello-brainstem dominant form of X-linked adrenoleukodystrophy presenting as multiple system atrophy: case report and literature review. Neuropathology 2016;36:64–76.ArticlePubMedPMC
- 86. Valianpour F, Selhorst JJ, van Lint LE, van Gennip AH, Wanders RJ, Kemp S. Analysis of very long-chain fatty acids using electrospray ionization mass spectrometry. Mol Genet Metab 2003;79:189–196.ArticlePubMed
- 87. Ognjenović J, Simonović M. Human aminoacyl-tRNA synthetases in diseases of the nervous system. RNA Biol 2018;15(4-5):623–634.ArticlePubMedPMC
- 88. Scheper GC, van der Klok T, van Andel RJ, van Berkel CG, Sissler M, Smet J, et al. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet 2007;39:534–539.ArticlePubMedPDF
- 89. Labauge P, Dorboz I, Eymard-Pierre E, Dereeper O, Boespflug-Tanguy O. Clinically asymptomatic adult patient with extensive LBSL MRI pattern and DARS2 mutations. J Neurol 2011;258:335–337.ArticlePubMedPDF
- 90. Lan MY, Chang YY, Yeh TH, Lin TK, Lu CS. Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL) with a novel DARS2 mutation and isolated progressive spastic paraparesis. J Neurol Sci 2017;372:229–231.ArticlePubMed
- 91. Tzoulis C, Tran GT, Gjerde IO, Aasly J, Neckelmann G, Rydland J, et al. Leukoencephalopathy with brainstem and spinal cord involvement caused by a novel mutation in the DARS2 gene. J Neurol 2012;259:292–296.ArticlePubMedPDF
- 92. Alexander WS. Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant. Brain 1949;72:373–381.3 pl. ArticlePubMed
- 93. Fu MH, Chang YY, Lin NH, Yang AW, Chang CC, Liu JS, et al. Recessively inherited adult-onset Alexander disease caused by a homozygous mutation in the GFAP gene. Mov Disord 2020;35:1662–1667.ArticlePubMedPDF
- 94. Russo LS Jr, Aron A, Anderson PJ. Alexander’s disease: a report and reappraisal. Neurology 1976;26:607–614.ArticlePubMed
- 95. Pareyson D, Fancellu R, Mariotti C, Romano S, Salmaggi A, Carella F, et al. Adult-onset Alexander disease: a series of eleven unrelated cases with review of the literature. Brain 2008;131(Pt 9):2321–2331.ArticlePubMed
- 96. Namekawa M, Takiyama Y, Honda J, Shimazaki H, Sakoe K, Nakano I. Adult-onset Alexander disease with typical “tadpole” brainstem atrophy and unusual bilateral basal ganglia involvement: a case report and review of the literature. BMC Neurol 2010;10:21.ArticlePubMedPMCPDF
- 97. Lamichhane A, Cabrero FR. Metachromatic leukodystrophy. [updated 2022 Sep 21]. In: StatPearls [Internet]. Treasure Island: StatPearls Publishing; c2022 [accessed on 2022 May 17]. Available at: https://www.ncbi.nlm.nih.gov/books/NBK560744/.
- 98. Gascon-Bayarri J, Campdelacreu J, Sánchez-Castañeda C, Martínez-Yélamos S, Moragas M, Scheper GC, et al. Leukoencephalopathy with vanishing white matter presenting with presenile dementia. J Neurol Neurosurg Psychiatry 2009;80:810–811.ArticlePubMed
- 99. Labauge P, Horzinski L, Ayrignac X, Blanc P, Vukusic S, Rodriguez D, et al. Natural history of adult-onset eIF2B-related disorders: a multicentric survey of 16 cases. Brain 2009;132(Pt 8):2161–2169.ArticlePubMed
- 100. van der Knaap MS, Pronk JC, Scheper GC. Vanishing white matter disease. Lancet Neurol 2006;5:413–423.ArticlePubMed
- 101. La Piana R, Vanderver A, van der Knaap M, Roux L, Tampieri D, Brais B, et al. Adult-onset vanishing white matter disease due to a novel EIF2B3 mutation. Arch Neurol 2012;69:765–768.ArticlePubMedPMC
- 102. Debs R, Froissart R, Aubourg P, Papeix C, Douillard C, Degos B, et al. Krabbe disease in adults: phenotypic and genotypic update from a series of 11 cases and a review. J Inherit Metab Dis 2013;36:859–868.ArticlePubMedPDF
- 103. Finnsson J, Sundblom J, Dahl N, Melberg A, Raininko R. LMNB1-related autosomal-dominant leukodystrophy: clinical and radiological course. Ann Neurol 2015;78:412–425.ArticlePubMedPMCPDF
- 104. Zhang Y, Li J, Bai R, Wang J, Peng T, Chen L, et al. LMNB1-related adult-onset autosomal dominant leukodystrophy presenting as movement disorder: a case report and review of the literature. Front Neurosci 2019;13:1030.ArticlePubMedPMC
- 105. Guo Z, Lu T, Peng L, Cheng H, Peng F, Li J, et al. CLCN2-related leukoencephalopathy: a case report and review of the literature. BMC Neurol 2019;19:156.ArticlePubMedPMCPDF
- 106. Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis 2010;5:16.ArticlePubMedPMCPDF
- 107. Sévin M, Lesca G, Baumann N, Millat G, Lyon-Caen O, Vanier MT, et al. The adult form of Niemann-Pick disease type C. Brain 2007;130(Pt 1):120–133.ArticlePubMed
- 108. Gburek-Augustat J, Groeschel S, Kern J, Beck-Woedl S, Just J, Harzer K, et al. Comparative analysis of cerebral magnetic resonance imaging changes in nontreated infantile, juvenile and adult patients with Niemann-Pick disease type C. Neuropediatrics 2020;51:37–44.ArticlePubMed
- 109. Srivastava S, Butala A, Mahida S, Richter J, Mu W, Poretti A, et al. Expansion of the clinical spectrum associated with AARS2-related disorders. Am J Med Genet A 2019;179:1556–1564.ArticlePubMedPDF
- 110. Gao J, Brackley S, Mann JP. The global prevalence of Wilson disease from next-generation sequencing data. Genet Med 2019;21:1155–1163.ArticlePubMedPDF
- 111. Leung M, Aronowitz PB, Medici V. The present and future challenges of Wilson’s disease diagnosis and treatment. Clin Liver Dis (Hoboken) 2021;17:267–270.ArticlePubMedPMCPDF
- 112. Lucena-Valera A, Perez-Palacios D, Muñoz-Hernandez R, Romero-Gómez M, Ampuero J. Wilson’s disease: revisiting an old friend. World J Hepatol 2021;13:634–649.ArticlePubMedPMC
- 113. Kumar N, Kumar D. Teaching video neuroimages: myoclonus as the presenting feature of Wilson disease. Neurology 2019;92:e1667–e1668.ArticlePubMed
- 114. Hitoshi S, Iwata M, Yoshikawa K. Mid-brain pathology of Wilson’s disease: MRI analysis of three cases. J Neurol Neurosurg Psychiatry 1991;54:624–626.ArticlePubMedPMC
- 115. Sánchez-Monteagudo A, Ripollés E, Berenguer M, Espinós C. Wilson’s disease: facing the challenge of diagnosing a rare disease. Biomedicines 2021;9:1100.ArticlePubMedPMC
- 116. van Wassenaer-van Hall HN. Neuroimaging in Wilson disease. Metab Brain Dis 1997;12:1–19.ArticlePubMedPDF
- 117. McNeill A, Chinnery PF. Neurodegeneration with brain iron accumulation. Handb Clin Neurol 2011;100:161–172.ArticlePubMed
- 118. Gregory A, Hayflick S. Neurodegeneration with brain iron accumulation disorders overview. [updated 2019 Oct 21]. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, et al. GeneReviews(R) [Internet]. Seattle: University of Washington; c1993-2023 [accessed on 2022 Apr 23]. Available at: https://www.ncbi.nlm.nih.gov/books/NBK121988/.
- 119. Hayflick SJ, Westaway SK, Levinson B, Zhou B, Johnson MA, Ching KH, et al. Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N Engl J Med 2003;348:33–40.ArticlePubMed
- 120. Salomão RP, Pedroso JL, Gama MT, Dutra LA, Maciel RH, Godeiro-Junior C, et al. A diagnostic approach for neurodegeneration with brain iron accumulation: clinical features, genetics and brain imaging. Arq Neuropsiquiatr 2016;74:587–596.ArticlePubMed
- 121. Kruer MC, Boddaert N. Neurodegeneration with brain iron accumulation: a diagnostic algorithm. Semin Pediatr Neurol 2012;19:67–74.ArticlePubMedPMC
- 122. Lee JH, Yun JY, Gregory A, Hogarth P, Hayflick SJ. Brain MRI pattern recognition in neurodegeneration with brain iron accumulation. Front Neurol 2020;11:1024.ArticlePubMedPMC
- 123. Min R, Depienne C, Sedel F, Abbink TEM, van der Knaap MS. CLCN2-related leukoencephalopathy [published 2015 Nov 5, updated 2021 May 20]. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, et al. GeneReviews(R) [Internet]. Seattle: University of Washington; c1993-2023 [accessed on 2022 Jul 11]. Available at: https://www.ncbi.nlm.nih.gov/books/NBK326661/.
- 124. Wei C, Qin Q, Chen F, Zhou A, Wang F, Zuo X, et al. Adult-onset vanishing white matter disease with the EIF2B2 gene mutation presenting as menometrorrhagia. BMC Neurol 2019;19:203.ArticlePubMedPMCPDF
- 125. Nardecchia F, Manti F, De Leo S, Carducci C, Leuzzi V. Clinical characterization of tremor in patients with phenylketonuria. Mol Genet Metab 2019;128(1-2):53–56.ArticlePubMed
- 126. Fine AS, Nemeth CL, Kaufman ML, Fatemi A. Mitochondrial aminoacyl-tRNA synthetase disorders: an emerging group of developmental disorders of myelination. J Neurodev Disord 2019;11:29.ArticlePubMedPMCPDF
- 127. Dallabona C, Diodato D, Kevelam SH, Haack TB, Wong LJ, Salomons GS, et al. Novel (ovario) leukodystrophy related to AARS2 mutations. Neurology 2014;82:2063–2071.ArticlePubMedPMC
- 128. Sundal C, Carmona S, Yhr M, Almström O, Ljungberg M, Hardy J, et al. An AARS variant as the likely cause of Swedish type hereditary diffuse leukoencephalopathy with spheroids. Acta Neuropathol Commun 2019;7:188.ArticlePubMedPMCPDF
- 129. Lynch DS, Zhang WJ, Lakshmanan R, Kinsella JA, Uzun GA, Karbay M, et al. Analysis of mutations in AARS2 in a series of CSF1R-negative patients with adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. JAMA Neurol 2016;73:1433–1439.ArticlePubMed
- 130. Członkowska A, Litwin T, Chabik G. Wilson disease: neurologic features. Handb Clin Neurol 2017;142:101–119.PubMed
- 131. Kono S. Aceruloplasminemia: an update. Int Rev Neurobiol 2013;110:125–151.PubMed
- 132. Yatziv S, Russell A. An unusual form of metachromatic leukodystrophy in three siblings. Clin Genet 1981;19:222–227.ArticlePubMed
- 133. Jansen AC, Andermann E, Niel F, Creveaux I, Boespflug-Tanguy O, Andermann F. Leucoencephalopathy with vanishing white matter may cause progressive myoclonus epilepsy. Epilepsia 2008;49:910–913.ArticlePubMed
- 134. Canafoglia L, Robbiano A, Pareyson D, Panzica F, Nanetti L, Giovagnoli AR, et al. Expanding sialidosis spectrum by genome-wide screening: NEU1 mutations in adult-onset myoclonus. Neurology 2014;82:2003–2006.ArticlePubMed
- 135. Guerreiro R, Kara E, Le Ber I, Bras J, Rohrer JD, Taipa R, et al. Genetic analysis of inherited leukodystrophies: genotype-phenotype correlations in the CSF1R gene. JAMA Neurol 2013;70:875–882.ArticlePubMedPMC
- 136. Bhatti A, Jain N, Desai K, Ravat SH, Agarwal PA. “Finger-Flapping” hand stereotypy as a presenting feature of Wilson’s disease. Mov Disord Clin Pract 2018;6:74–76.ArticlePubMedPMCPDF
- 137. Rauschka H, Colsch B, Baumann N, Wevers R, Schmidbauer M, Krammer M, et al. Late-onset metachromatic leukodystrophy: genotype strongly influences phenotype. Neurology 2006;67:859–863.ArticlePubMed
- 138. Schneider SA, Dusek P, Hardy J, Westenberger A, Jankovic J, Bhatia KP. Genetics and pathophysiology of Neurodegeneration with Brain Iron Accumulation (NBIA). Curr Neuropharmacol 2013;11:59–79.ArticlePubMedPMC
- 139. Sim CY, Mukari SAM, Ngu LH, Loh CY, Remli R, Ibrahim NM. Labrune’s syndrome presenting with stereotypy-like movements and psychosis: a case report and review. J Mov Disord 2022;15:162–166.ArticlePubMedPMCPDF
- 140. Labrune P, Lacroix C, Goutières F, de Laveaucoupet J, Chevalier P, Zerah M, et al. Extensive brain calcifications, leukodystrophy, and formation of parenchymal cysts: a new progressive disorder due to diffuse cerebral microangiopathy. Neurology 1996;46:1297–1301.ArticlePubMed
- 141. Osman O, Labrune P, Reiner P, Sarov M, Nasser G, Riant F, et al. Leukoencephalopathy with calcifications and cysts (LCC): 5 cases and literature review. Rev Neurol (Paris) 2020;176:170–179.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
Comments on this article
JEE YOUNG LEE Cancel
 PubReader
PubReader ePub Link
ePub Link Cite
Cite
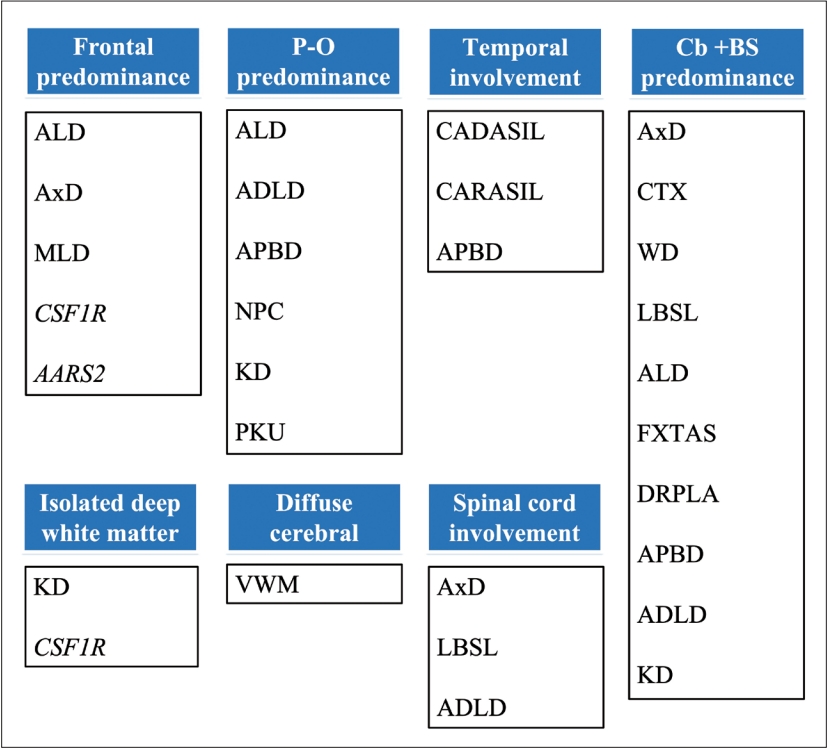
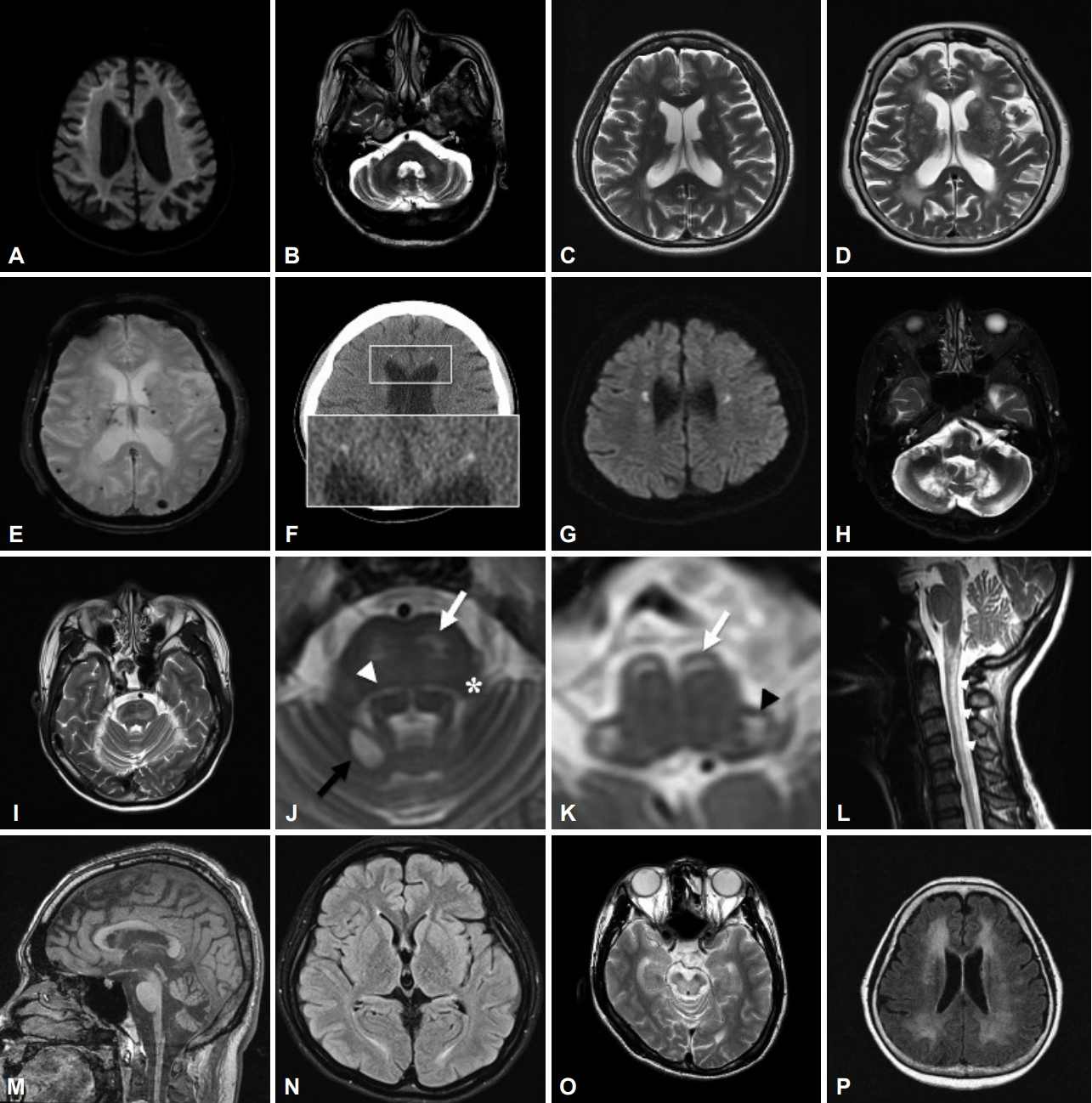


JEE YOUNG LEE
August 04, 2023