E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 16(3); 2023 > Article
-
Review Article
GBA1 Variants and Parkinson’s Disease: Paving the Way for Targeted Therapy -
Young Eun Huh1
 , Tatiana Usnich2, Clemens R. Scherzer3,4, Christine Klein2
, Tatiana Usnich2, Clemens R. Scherzer3,4, Christine Klein2 , Sun Ju Chung5
, Sun Ju Chung5 -
Journal of Movement Disorders 2023;16(3):261-278.
DOI: https://doi.org/10.14802/jmd.23023
Published online: June 12, 2023
1Department of Neurology, CHA Bundang Medical Center, CHA University, Seongnam, Korea
2Institute of Neurogenetics, University of Lübeck and University Hospital of Schleswig-Holstein, Lübeck, Germany
3Advanced Center for Parkinson’s Disease Research, Harvard Medical School, Brigham and Women’s Hospital, Boston, MA, USA
4Precision Neurology Program, Harvard Medical School, Brigham and Women’s Hospital, Boston, MA, USA
5Department of Neurology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
- Corresponding author: Sun Ju Chung, MD, PhD Department of Neurology, Asan Medical Center, University of Ulsan College of Medicine, 88 Olympic-ro 43-gil, Songpa-gu, Seoul 05505, Korea / Tel: +82-2-3010-3440 / Fax: +82-2-474-4691 / E-mail: sjchung@amc.seoul.kr
- Corresponding author: Christine Klein, MD Institute of Neurogenetics, University of Lübeck and University Hospital of Schleswig-Holstein, BMF, Building 67, Room 067.000 10 047.00, Ratzeburger Allee 160, Campus Lübeck, Lübeck 23538, Germany / Tel: +49-451-3101-8200 / Fax: +49-451-3101-8204 / E-mail: christine.klein@neuro.uniluebeck.de
Copyright © 2023 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
ABSTRACT
- Glucosylceramidase beta 1 (GBA1) variants have attracted enormous attention as the most promising and important genetic candidates for precision medicine in Parkinson’s disease (PD). A substantial correlation between GBA1 genotypes and PD phenotypes could inform the prediction of disease progression and promote the development of a preventive intervention for individuals at a higher risk of a worse disease prognosis. Moreover, the GBA1-regulated pathway provides new perspectives on the pathogenesis of PD, such as dysregulated sphingolipid metabolism, impaired protein quality control, and disrupted endoplasmic reticulum-Golgi trafficking. These perspectives have led to the development of novel disease-modifying therapies for PD targeting the GBA1-regulated pathway by repositioning treatment strategies for Gaucher’s disease. This review summarizes the current hypotheses on a mechanistic link between GBA1 variants and PD and possible therapeutic options for modulating GBA1-regulated pathways in PD patients.
- Parkinson’s disease (PD) is a neurodegenerative disease with multifactorial etiologies resulting from a complex interplay of genetic and environmental factors. Although the etiology of PD has been considered mostly idiopathic, recent studies have suggested a strong influence of genetic factors on the risk of PD, with a heritability of up to 30% [1,2]. Among 90 susceptibility loci linked to the risk of PD [3], glucosylceramidase beta 1 (GBA1) variants, the most common genetic risk factor, have recently attracted much attention.
- The GBA1 gene, located on chromosome 1q21, encodes the lysosomal enzyme β-glucocerebrosidase, which hydrolyzes glucosylceramide into glucose and ceramide [4]. Initially, homozygous or compound heterozygous variants in the GBA1 gene were noted to cause Gaucher’s disease (GD), the most common lysosomal storage disorder [5,6]. More than 300 genetic variants in the GBA1 gene have been associated with GD, including missense mutations, frameshifts, insertions, deletions, and complex alleles [7,8]. The most common GD-causing genetic variants are the point mutations p.N409S (N370S) and p.L483P (L444P). In GD, β-glucocerebrosidase activity is markedly reduced, resulting in the accumulation of its direct substrate, glucosylceramide, in macrophages, forming Gaucher cells [6]. The lysosomal accumulation of glucocerebroside results in multisystem disorders, including hepatosplenomegaly, anemia, thrombocytopenia, bone disease, and neurological manifestations. GD is traditionally categorized into 3 clinical subtypes based on the absence (nonneuropathic, type I) or presence (neuropathic, types II and III) of central nervous system (CNS) involvement. In GD, genotypephenotype relationships have been relatively well described, i.e., the p.N409S variant is exclusively associated with nonneuropathic GD, while the p.L483P variant is generally associated with neuropathic disease [6].
- Here, we review the current understanding of the role of GBA1 variants in the development of PD. We also discuss the molecular basis underlying the link between GBA1 variants and PD pathology, and we assess a new therapeutic approach for PD targeting GBA1-modulating pathways.
INTRODUCTION
- Whereas homozygous or compound heterozygous GBA1 variants cause GD, heterozygous or several compound heterozygous/ homozygous GBA1 variants increase the risk for PD. The relationship between GBA1 variants and PD was initially reported in a patient with type 1 GD who presented typical Parkinsonian signs [9]. Heterozygous GBA1 variant carriers who are relatives of GD patients are at an increased risk of developing PD [10]. In a large meta-analysis of patients from 16 centers, including individuals of different ethnicities, an estimated odds ratio (OR) for carrying a GBA1 variant among PD patients of 5.43 was found across the cohorts [7]. Initially, genome-wide association studies (GWASs) failed to identify GBA1 as a susceptibility gene for PD [11-13]. Reasons for this include the presence of several rare variants with incomplete penetrance or the occurrence of different haplotypes [14]. Ensuing large-scale GWASs eventually confirmed that a GBA1 variant was a strong risk factor for PD by examining specific GBA1 variants [15-19].
- Although the presence of a GBA1 variant is the most common genetic risk factor for PD, the frequency of GBA1 variants in PD patients varies depending on ethnicity and sequencing methods. GBA1 variants are particularly frequent in PD patients of Ashkenazi Jewish (AJ) ancestry, with a frequency of up to 31.3% [20,21], while approximately 2%–10% of European or North American non-AJ PD patients carry a GBA1 variant [22-25]. Among northeastern Asian patients with PD, the proportion of those carrying GBA1 variants is 2%–10.7% among Chinese patients [26-28], 3.2% among South Korean patients [29], and 9.4% among Japanese patients [30]. To date, approximately 130 heterozygous GBA1 variants are thought to be associated with an increased PD risk [31].
- Although penetrance is typically estimated in a monogenic disease caused by rare and highly pathogenic variants with Mendelian inheritance, age-dependent lifetime penetrance can also be analyzed in a common complex disease, such as PD, using population allele frequencies [32]. The age-dependent lifetime penetrance of GBA1 variants is relatively low. Up to 1% of individuals in the general population are heterozygous GBA1 variant carriers [33], and the frequency increases up to 8% in the AJ population [9]. Two prospective studies showed that the age-specific cumulative risk of PD among GBA1 heterozygotes is 1.5%–2.2% by ages 60–65 years and 7.7%–10.9% by ages 80–85 years [34,35]. Recently, the penetrance of the four most common PD-associated GBA1 variants, including p.E365K (E326K), p.T408M (T369M), p. N409S, and p.L483P, was estimated in a large cohort of 3,832 unrelated Italian patients with PD and 7,757 healthy controls [32]. Notably, the penetrance of p.L483P reached 15.07% above age 75 years, with 5.42% by age 75 years and 1.40% by age 65 years. The penetrance of the other three variants above age 75 years was estimated as 2.15% for p.T408M, 2.96% for p.E365K, and 4.72% for p.N409S.
GBA1 VARIANTS AND PD RISK
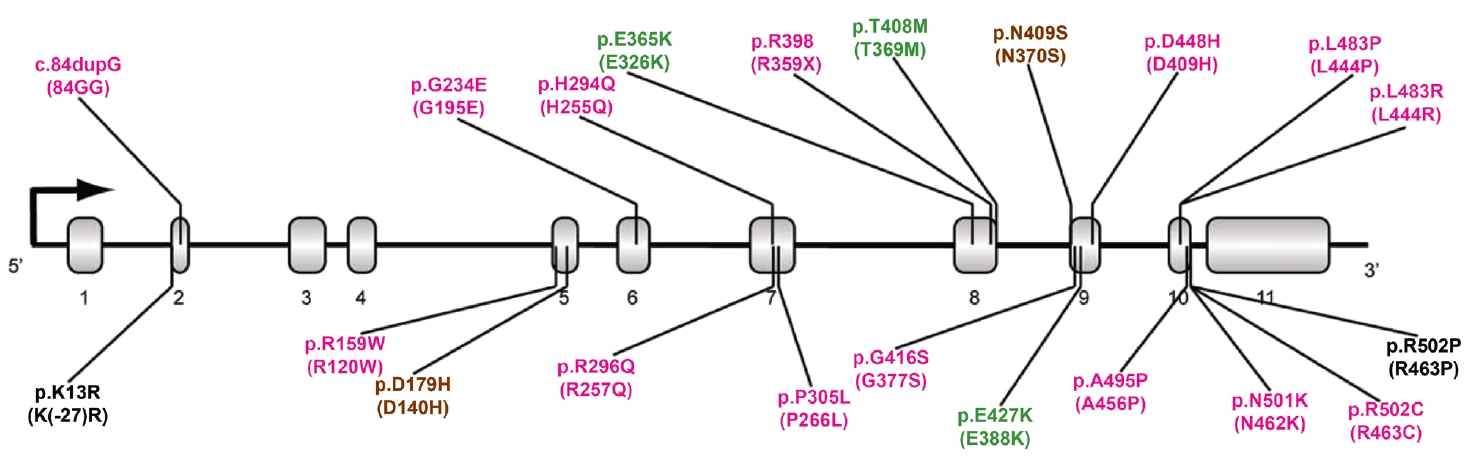
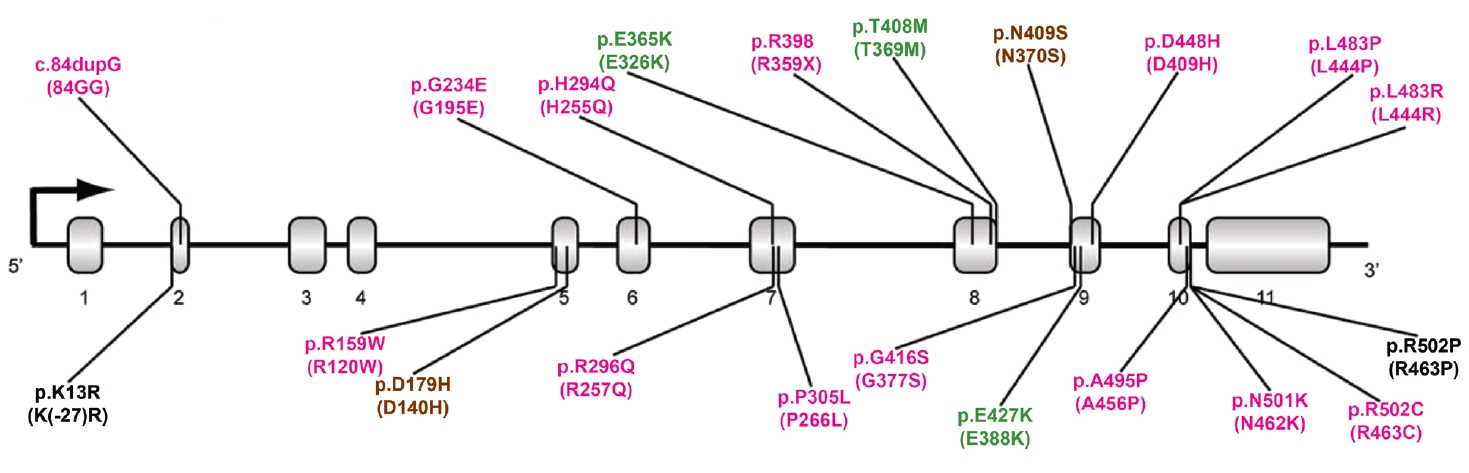
- More than 100 GBA1 variants have been associated with PD. GBA1 variants can be classified as severe, mild, and risk variants based on their historical association with variants causing GD (neuropathic or nonneuropathic) or their association with PD risk and phenotypic severity (Figure 1) [36]. Severe variants are associated with neuropathic GD, such as p.L483P, p.L483R (L444R), p.A495P (A456P), and p.R159W (R120W). These GBA1 variants are reported to markedly increase the risk for PD and accelerate disease progression in PD patients. The heterozygous carriers of mild variants include PD patients with GBA1 variants, such as p.N409S, which causes nonneuropathic GD. In some studies, the disease risk and the rate of disease progression were found to be moderately elevated in this group. A detailed description is given in the “GBA1 variants and PD phenotypes” section. In addition to the known pathogenic variants causing GD, many studies have discovered GBA1 variants that are not pathogenic for GD but predispose to PD (risk variants), including p.E365K, p.T408M, and p.E427K (E388K). p.E365K is associated with an increased risk for PD [7,17] and an earlier age at onset [37,38]. Longitudinal studies show that GBA1 risk variants, such as p.E365K, p.T408M, and p.E427K, predict more rapid motor and cognitive progression [39,40]. A recent meta-analysis demonstrated that the p.E365K variant increases the risk of developing cognitive impairment (3-fold) among PD patients whose cognition was normal at baseline. At the same time, p.T408M is associated with faster progression of motor disability [41]. These variants are also associated with the development of rapid eye movement (REM) sleep behavior disorder (RBD) [41].
TYPES OF GBA1 VARIANTS IN PD
- Neuropathologically, PD patients with a GBA1 variant (GBA1-PD patients) are indistinguishable from those with idiopathic PD, exhibiting nigral dopaminergic loss and Lewy body pathology [42,43]. The phenotype of GBA1-PD patients appears similar to that of idiopathic PD patients at an individual level. However, increasing evidence from recent clinical studies indicates that GBA1 variant carriers present a more aggressive disease manifestation (Table 1).
- In GBA1-PD patients, the age at onset is 3–6 years earlier (mean of 50–55 years) [42,44-52], and the mortality risk is increased (hazard ratio = 1.85) compared to idiopathic PD patients [46]. Motor disability is more severe, and progression to Hoehn and Yahr stage 3 is faster in GBA1-PD patients than in idiopathic PD patients [39,46,52-54]. Freezing of gait, dysphagia, and dysarthria are more frequent in GBD1-PD patients [46], while levodopa responsiveness is comparable to that in idiopathic PD patients [42,46,49,55,56]. GBA1 variants are associated with a 2.4- to 3-fold greater risk of dementia [46,57] and an earlier age at the onset of cognitive impairment [25,46,54,58,59]. The rate of cognitive decline is much faster in GBA1-PD patients than in noncarriers of pathogenic GBA1 variants [25,40,46]. Visual hallucinations are more frequent [42,46,49,53,59,60] and occur at a younger age in GBA1-PD patients [42,46,59]. Other neuropsychiatric symptoms, including depression, anxiety, and apathy, are more prevalent in GBA1-PD patients than in idiopathic PD patients [42,46,57,60,61]. GBA1-PD patients have a higher risk of autonomic dysfunction, such as orthostatic hypotension, constipation, and urinary/sexual dysfunction [46,60,62]. RBD is 2- to 3-fold more common in GBA1-PD patients than in idiopathic PD patients [49,63]. In idiopathic RBD patients, GBA1 variants are more frequent than in healthy controls [63-65]. Moreover, idiopathic RBD patients with a GBA1 variant exhibit a higher risk for phenoconversion to PD than those without a GBA1 variant [65].
- GBA1 variant status may also affect presynaptic striatal dopaminergic integrity. GBA1-PD patients show a more profound reduction in dopamine transporter binding than idiopathic PD patients [46,66,67]. Greater asymmetry in striatal binding is observed in GBA1-PD patients than in idiopathic PD patients as well as PD patients carrying SNCA, PINK1, or PARK2 variants [66,68]. A longitudinal study demonstrated more marked 123I-FP-CIT SUVr reduction in the striatal and extrastriatal regions in GBA1-PD patients than in idiopathic PD patients [69]. In this study, 123I-FP-CIT SUVr reduction in idiopathic PD patients became similar to that in GBA1-PD patients over time, suggesting that GBA1 variants accelerate the early neurodegenerative processes in PD, leading to a malignant clinical phenotype. In contrast, de novo GBA1-PD patients enrolled in the Parkinson’s Progression Markers Initiative cohort exhibited less severe presynaptic striatal dopaminergic loss in dopamine transporter images than idiopathic PD patients [70]. Another study showed that despite similar levels of presynaptic striatal dopaminergic availability, GBA1-PD patients exhibited more severe motor disability on the less affected side than idiopathic PD patients, indicating a detrimental effect of GBA1 variants on motor reserve [71].
- Interestingly, GBA1 variants causing neuropathic GD (severe GBA1 variants) are associated with even more severe PD phenotypes than those related to nonneuropathic GD (mild GBA1 variants), suggesting a genotype-phenotype correlation in GBA1-PD patients. GBA1-PD patients with a severe variant show an increased risk for developing PD [45,72] and a younger age at onset than those with mild variants [42,46,51]. The genotype-phenotype relationship in GBA1-PD patients has been well described regarding cognitive impairment. GBA1-PD patients with a severe variant show an increased risk for dementia up to 3-fold and a more rapid cognitive progression than those with mild variants [46]. Severe GBA1 variants or complex alleles specifically accelerate cognitive decline among PD patients compared to the wild type [25,49]. Motor disability is more severe, and the progression is faster in GBA1-PD patients with a severe variant than in noncarriers [49,52,59]. On dopamine transporter imaging, a more profound loss of presynaptic dopaminergic terminals is observed in GBA1-PD patients with a severe variant than in those with a mild variant [46]. These data indicate that GBA1 genotypes are substantially correlated with the severity of the PD phenotype in a variant dose-dependent manner.
GBA1 VARIANTS AND PD PHENOTYPES
- Increasing evidence suggests that β-glucocerebrosidase links GBA1 variants with the accumulation of α-synuclein primarily through a loss-of-function mechanism. In vivo and in vitro studies showed that reducing the enzymatic activity of β- glucocerebrosidase induces the accumulation of α-synuclein similar to that observed in the brains of GD or PD subjects [73-77]. The knockdown of wild-type β-glucocerebrosidase in primary neurons using shRNA causes a dramatic increase in the level of oligomeric α-synuclein [73]. The enzymatic inhibition of β- glucocerebrosidase by conduritol B epoxide elevates α-synuclein aggregation in human neuroblastoma cells [74] and the substantia nigra (SN) of mice [75]. α-Synuclein and ubiquitin aggregates accumulate in the brains of mice harboring homozygous GBA1 variants, such as p.D448H (D409H) or p.V433L (V394L) [76]. Midbrain dopaminergic neurons derived from the induced pluripotent stem cells (iPSCs) of PD patients carrying a heterozygous GBA1 variant (p.L483P or p.N409S) had reduced β- glucocerebrosidase activity and an increase in the levels of glucosylceramide and α-synuclein [77]. However, some in vitro study findings contradicted the loss-of-function mechanism by demonstrating the accumulation of α-synuclein without a reduction in β- glucocerebrosidase activity in GBA1 mutant-overexpressing neuronal cell lines (p.N409S or p.L483P) [78]. In addition, a pathology study did not confirm substrate accumulation in the presence of reduced β-glucocerebrosidase function in the brains of neuropathic GD or PD patients carrying a heterozygous GBA1 variant [79]. Accordingly, the level of β-glucocerebrosidase activity in GBA1-associated and idiopathic PD patients has been under intense investigation as a first step to elucidate a mechanistic link between GBA1 variants and PD and to validate its possible utility as a biomarker of GBA1-PD patients.
- In GBA1-PD patients, β-glucocerebrosidase activity is generally reduced in brain, cerebrospinal fluid (CSF), and peripheral blood samples. In the brain regions of GBA1-PD patients, including the SN, putamen, cerebellum, and amygdala, β- glucocerebrosidase activity was significantly reduced compared to healthy controls [80]. The protein level of β-glucocerebrosidase was found to be reduced in the SN, putamen, and cerebellum, while the mRNA expression did not differ from healthy controls. A recent brain autopsy study also showed that β-glucocerebrosidase activity in the SN, putamen and frontal cortex of GBA1-PD patients carrying a mild or severe GBA1 variant was diminished compared to that in idiopathic PD patients and healthy controls [81]. However, the β-glucocerebrosidase activity in the SN, putamen, and frontal cortex of GBA1-PD patients carrying a GBA1 risk variant did not differ from that in idiopathic PD patients, but it was reduced compared to that in healthy controls. In the CSF of GBA1-PD patients, β-glucocerebrosidase activity is lower than in that of noncarriers [82,83]. β-Glucocerebrosidase activity in peripheral blood samples, including dried blood spots from frozen whole blood [84-86], dried blood spots from fresh whole blood [87], and peripheral blood mononuclear cells [88,89], is also reduced in GBA1-PD patients, similar to that observed in the brain and CSF. A recent study showed that the severity of GBA1 variants (severe, mild, and risk variants) is negatively correlated with β- glucocerebrosidase activity in a quantitative manner [85]. In this study, the more severe GBA1 variant types showed a more rapid decrease in β-glucocerebrosidase activity over time. This may suggest that blood β-glucocerebrosidase activity is a quantitative endophenotype that can be monitored noninvasively and targeted therapeutically.
- Notably, a reduction in β-glucocerebrosidase activity has also been reported in biosamples from PD patients without a GBA1 variant. In the brains of idiopathic PD patients, β- glucocerebrosidase activity was reduced in the SN and cerebellum compared to healthy controls [80]. The protein level of β- glucocerebrosidase was also reduced in the SN and cerebellum [80], while the mRNA level was comparable [80] or decreased [90]. The enzymatic activity and protein levels of β-glucocerebrosidase in the anterior cingulate gyrus were lower in both early- and late-stage idiopathic PD patients than in healthy controls [91]. This study found a negative correlation between β-glucocerebrosidase activity and α-synuclein levels in the anterior cingulate cortex. In the CSF of idiopathic PD patients, β-glucocerebrosidase activity was reduced compared to that of healthy controls [83,92]. The diagnostic accuracy of CSF β-glucocerebrosidase activity for PD was high, with a specificity of 77% and a sensitivity of 67% [82]. In contrast, there have been contradictory results for β- glucocerebrosidase activity in the peripheral blood of idiopathic PD patients. A sizeable single-center study reported that β- glucocerebrosidase activity in the dried blood spots from fresh whole blood of idiopathic PD patients was reduced by 5% compared to that of healthy controls [87]. However, other studies did not find any difference in β-glucocerebrosidase activity in dried blood spots from frozen whole blood or peripheral blood mononuclear cells between idiopathic PD patients and healthy controls [85,86,88,89,93]. A longitudinal study of the changes in blood β-glucocerebrosidase activity in idiopathic PD patients found that they did not significantly differ from those of healthy controls [85]. A recent study measuring β-glucocerebrosidase activity in the dried blood spots of frozen whole blood samples from de novo idiopathic PD patients also did not show any difference between idiopathic PD patients and healthy controls [84]. These discrepancies may result from preanalytical variables, including sample storage, cohort composition, and assay methods.
- Many clinical studies collectively show reduced β- glucocerebrosidase activity in GBA1-PD patients, supporting the loss-of-function mechanism. Therefore, a significant reduction in the enzyme activity or protein levels of β-glucocerebrosidase provides a plausible rationale for β-glucocerebrosidase augmentation therapy for GBA1-PD or idiopathic PD [94,95]. However, a toxic gain-of-function hypothesis may also play a role. Indeed, α-synuclein aggregates were found to accumulate in the brains of heterozygous GBA1 knock-in (GBA1D409V/+) mice but not in those of heterozygous GBA1 knock-out (GBA1+/-) mice, while residual β-glucocerebrosidase activity was similarly reduced in both GBA1D409V/+ and GBA1+/- mice [96]. This finding indicates that a single dose of mutant β-glucocerebrosidase (D409V) may also contribute to the accumulation of α-synuclein aggregates through a toxic gain-of-function mechanism. Another issue that needs to be addressed is low β-glucocerebrosidase activity in idiopathic PD patients without a GBA1 variant, which implies a possible bidirectional interaction between β-glucocerebrosidase and α-synuclein. These issues will be discussed in the next section, “Pathophysiology leading to treatment avenues.”
β-GLUCOCEREBROSIDASE ACTIVITY IN GBA1-ASSOCIATED AND IDIOPATHIC PD
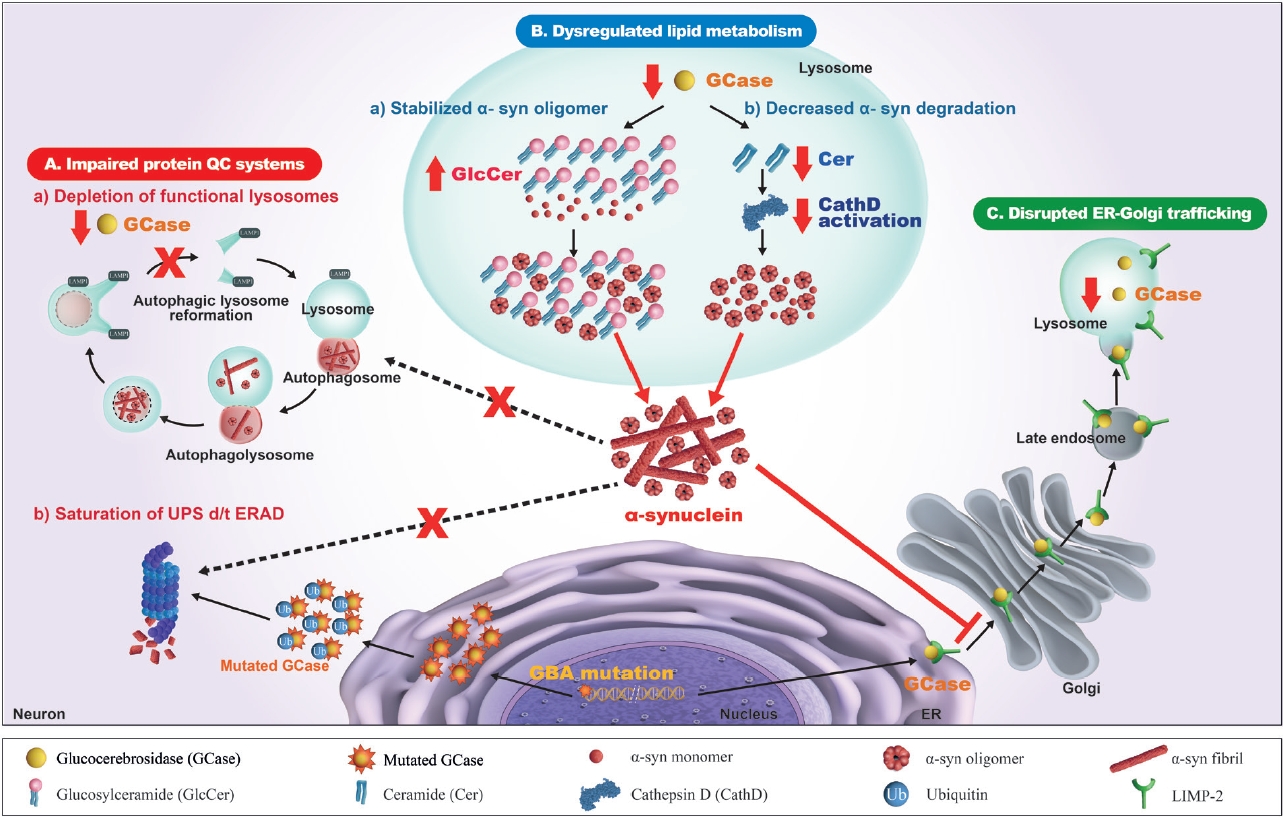
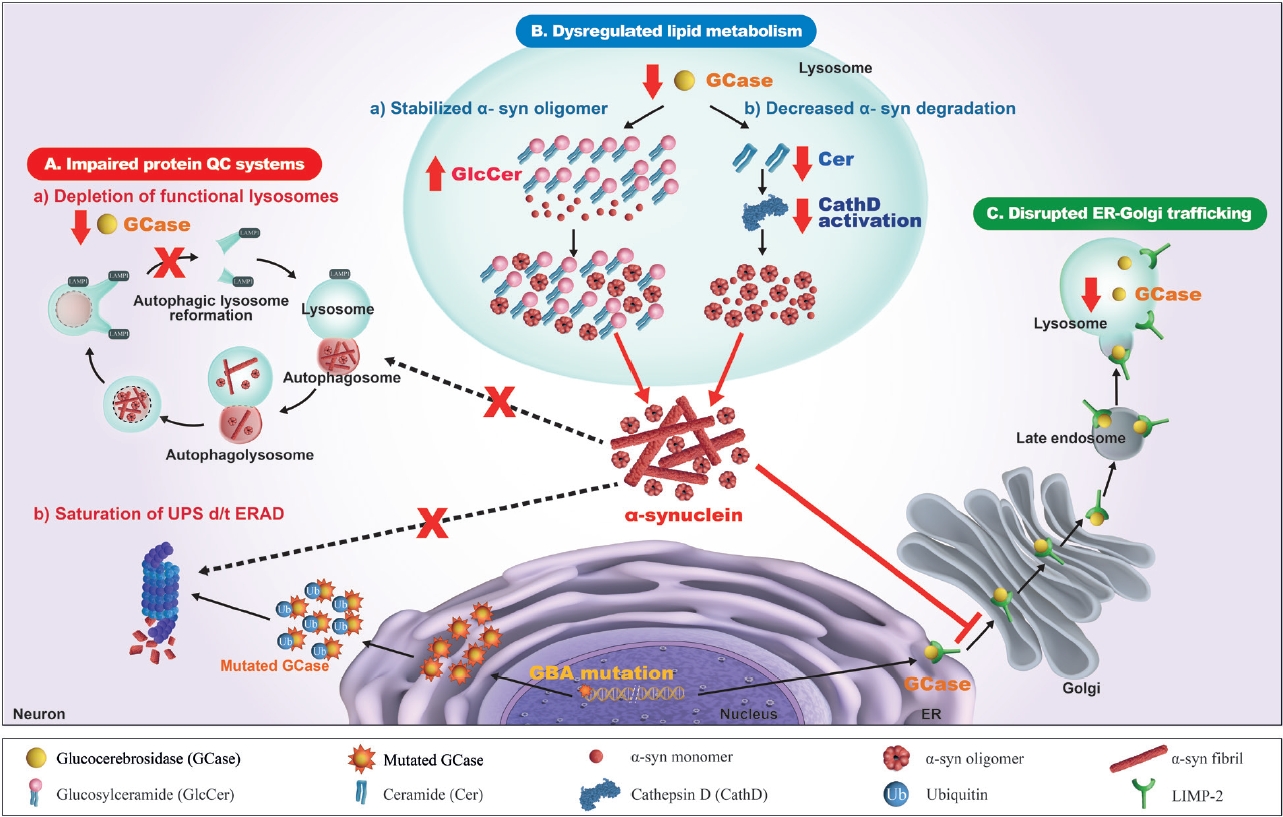
- There have been numerous hypotheses on the molecular mechanism linking β-glucocerebrosidase and α-synuclein in PD, but to date, none of them is supported by clinical and experimental data (Figure 2).
- Impaired protein quality control systems
- It is well accepted that one of the major mechanisms of the pathogenesis of PD is an impaired autophagy-lysosomal pathway, which may be the basic mechanism for new therapeutic development. Numerous PD-associated genes are involved in this system, including GBA1, LRRK2, SNCA, VPS35, SCARB2, and CTSD [97]. Alterations in β-glucocerebrosidase and other lysosomal enzymes, such as cathepsin D and a-galactosidase A, are observed in GBA1-PD and idiopathic PD patients [82,83,98,99]. Accordingly, an impaired autophagy-lysosomal pathway is ascribed to the crosstalk between β-glucocerebrosidase and α-synuclein. In dopaminergic neurons derived from the iPSCs of PD patients carrying heterozygous GBA1 variants (p.L483P or p.N409S), an autophagy-lysosomal defect is detected together with reduced β-glucocerebrosidase activity, an accumulation of glucosylceramide and α-synuclein, and perturbed calcium homeostasis [77]. An autophagy-lysosomal disturbance is observed in relation to ER stress in dopaminergic neurons differentiated from iPSCs of PD patients carrying a heterozygous p.N409S variant [100]. GBA1 knockdown inhibits autophagy and accumulates α-synuclein via the inactivation of protein phosphatase 2A (PP2A) in neuroblastoma cells and rat primary cortical neurons, which is reversed by a PPP2A agonist [101]. β-Glucocerebrosidase deficiency inhibits autophagy-lysosomal reformation and causes the depletion of functional lysosomes in fibroblasts derived from PD patients with GBA1 variants [102]. Mutant β-glucocerebrosidase also impairs chaperone-mediated autophagy (CMA). In postmortem human GBA1-PD brains, half of the mutant β-glucocerebrosidase is mislocalized on the lysosomal surface [103]. This mislocalization depends on a pentapeptide motif in β-glucocerebrosidase used to target the cytosolic protein for degradation by CMA. Accordingly, mutant β-glucocerebrosidase on the lysosomal surface inhibits CMA, causing an accumulation of CMA substrates, including α-synuclein.
- In addition to the impaired autophagy-lysosomal pathway, several experimental studies have shown that GBA1 variants perturb the ubiquitin‒proteasome system (UPS), causing αsynuclein accumulation. Misfolded mutant β- glucocerebrosidase protein fails to be transported from the endoplasmic reticulum (ER) to lysosomes and is retained in the ER. This subsequently triggers ER-associated degradation (ERAD) [104], which is normally processed by UPS [105,106]. However, excessive ERAD saturates UPS and blocks UPS function to respond to the accumulation of other proteins, such as α-synuclein [107-109]. These findings favor a toxic gain-of-function mechanism, although other experiments could not replicate the ERAD/UPS activation induced by GBA1 variants [78,110,111].
- Dysregulated sphingolipid metabolism
- As β-glucocerebrosidase regulates sphingolipid metabolism, it has been suggested that an alteration in the profile of sphingolipids, such as glucosylceramide and ceramide, may contribute to crosstalk between β-glucocerebrosidase and α-synuclein. This pathogenic mechanism may provide useful information for the development of new therapeutics for PD.
- In vitro studies showed that accumulated glucosylceramide stabilizes the soluble oligomeric intermediate form of α- synuclein and promotes the aggregation of α-synuclein [73,112]. Glucosylceramide directly converts high molecular weight physiological α-synuclein conformers into assembly state intermediates in cell-free in vitro models without disassembly into free monomers [113]. This toxic conversion is reversed by treatment with a glucosylceramide synthase inhibitor in dopaminergic neurons derived from the iPSCs of PD patients with and without GBA1 variants. Another in vitro study using dopaminergic neurons derived from GBA1-PD patients carrying the p.N409S variant showed that the accumulation of intracellular glucosylceramide destabilizes normal α-synuclein tetramers/multimers and induces soluble toxic α-synuclein assemblies, which is reversed by reducing glucosylceramide [114]. In a mouse model of synucleinopathy, treatment with a glucosylceramide synthase inhibitor alleviates α-synuclein pathology and restores normal behavior [115]. This direct interaction between glucosylceramide and α- synuclein explains the specific accumulation of α-synuclein caused by β-glucocerebrosidase deficiency. However, this hypothesis has been debated because an accumulation of glucosylceramide has not been observed in the postmortem brain tissue or plasma of GBA1-PD patients [79,116].
- A recent study testing the sphingolipid profile in the CSF of drug-naïve early-stage PD patients showed that the glucosylceramide proportion is significantly increased in GBA1-PD patients [117]. In this study, the CSF glucosylceramide proportion was significantly correlated with the level of CSF α-synuclein. These findings strongly support β-glucocerebrosidase substrate reduction as a therapy for GBA1-PD or idiopathic PD patients [115,118]. Alternatively, glucosylceramide may be a bystander of glucosylsphingosine, the other substrate of β-glucocerebrosidase, because the accumulation of glucosylsphingosine, rather than glucosylceramide, initiated the aggregation of oligomeric α-synuclein in the brains of young GD/PD mice [119]. Consistent with this report, GBA1 haploinsufficiency accelerated α-synuclein pathology in α-synuclein transgenic mice, in which the level of glucosylsphingosine, not that of glucosylceramide, was elevated [120].
- Alterations in ceramide, the downstream product of β- glucocerebrosidase, have also been considered a possible link between the reduced β-glucocerebrosidase activity and α- synuclein formation in PD. Ceramide normally binds to lysosomal cathepsin D and activates cathepsin D into a catalytic form [121,122]. As cathepsin D directly degrades monomeric and aggregated α-synuclein in lysosomes [123-127], a reduction in ceramide caused by a loss of β-glucocerebrosidase function may impede the activation of cathepsin D and impair α-synuclein degradation [128]. In β-glucocerebrosidase-deficient cells, the inhibition of acid ceramidase restores the decreased level of ceramide and alleviates α-synuclein aggregation [128]. Variants of SMPD1 [129-132] encoding acid sphingomyelinase and GALC (galactosylceramidase) [19] are considered susceptibility genes for PD and other synucleinopathies. In the brains of idiopathic PD patients, the ceramide level is reduced in association with impaired β-glucocerebrosidase function and the accumulation of α-synuclein [91,133]. CSF levels of ceramide and its byproducts are also altered in GBA1-PD patients, probably through alternative processing of glucosylceramide: lysosomal glucosylceramides and glucosylsphingosines exit from the lysosome into the cytosol, where they are hydrolyzed by cytosolic β-glucocerebrosidase 2, resulting in increased levels of ceramide, sphingosine, and sphingosine-1-phosphate [134]. The plasma ceramide level is increased in both GBA1-PD [116] and idiopathic PD patients [135].
- In neurons derived from PD patients carrying either GBA1 or SNCA variants, altered sphingolipid metabolism shifts the physiological α-synuclein tetramer-monomer equilibrium to aggregation-prone monomers [114,136]. Conversely, the overexpression of β-glucocerebrosidase increases the α-synuclein tetramer-monomer ratio, reduces lipid-rich α-synuclein aggregates, and ameliorates PD-like phenotypes in mice [137]. Furthermore, the sphingolipid profile of fibroblasts derived from PD patients with the p.L483P variant shows an overall increase in sphingolipid levels with a higher proportion of short-chain sphingolipids, which are correlated with lower β-glucocerebrosidase activity [138]. Moreover, lipid extracts from these fibroblasts accelerate the aggregation of α-synuclein aggregates, which is reversed by ambroxol treatment. These findings all support a direct relationship between sphingolipids and α-synuclein accumulation in PD pathogenesis.
- Disrupted ER-to-Golgi trafficking of β- glucocerebrosidase through α-synuclein accumulation
- β-Glucocerebrosidase is usually transported from the ER to the Golgi, and then it reaches the lysosome, mediated by lysosomal integral membrane protein-2 (LIMP-2) [139]. Several lines of evidence from experimental and clinical studies indicate that ER-Golgi trafficking of β-glucocerebrosidase is blocked by the accumulation of α-synuclein, which leads to a reduction in βglucocerebrosidase in the lysosomal compartment [140]. The overexpression of α-synuclein in primary cortical neurons induces an accumulation of the ER form of β-glucocerebrosidase but a decrease in its post-ER form, which reduces lysosomal β- glucocerebrosidase activity [73,141]. In the postmortem brain cortex of a patient carrying the SNCA variant and idiopathic PD patients, the ER form of β-glucocerebrosidase was found to be elevated, while the ratio of post-ER to ER forms was reduced [141]. α- Synuclein knockdown restores ER-Golgi trafficking and β- glucocerebrosidase activity in neuronal synucleinopathy models derived from PD patients carrying SNCA or GBA1 variants and idiopathic PD patients [142]. In this study, α-synuclein accumulation blocked vesicular fusion at the cis-Golgi by causing aberrant associations with cis-Golgi-tethering factor GM130 and inducing alterations in the ER-Golgi localization of rab1a. The overexpression of rab1a restores the trafficking of β-glucocerebrosidase and decreases α-synuclein accumulation. These findings have prompted testing of the therapeutic efficacy of small molecule chaperones (SMCs) that enhance the ER-Golgi trafficking of β-glucocerebrosidase [143-147].
- In addition to the blockade of the ER-Golgi trafficking of β-glucocerebrosidase, α-synuclein can directly interact with β-glucocerebrosidase at lysosomal pH [148]. An in vitro study showed that β-glucocerebrosidase, which is partially inserted into the lysosomal membrane, typically binds to α-synuclein embedded in the lipid bilayer of lysosomes [149]. The authors postulated that the interaction between β-glucocerebrosidase and α-synuclein displaces β-glucocerebrosidase away from the membrane, possibly impeding its access to the substrate and perturbing the active site of β-glucocerebrosidase.
- Collectively, these data support an inverse relationship between β-glucocerebrosidase deficiency and α-synuclein accumulation, which explains the reduced β-glucocerebrosidase activity observed in PD patients in the absence of GBA1 variants. Moreover, this bidirectional interaction between β- glucocerebrosidase and α-synuclein further induces the accumulation of αsynuclein, leading to a vicious cycle for the propagation of disease pathology.
- The role of extracellular α-synuclein secretion
- Experimental studies have reported that β- glucocerebrosidase deficiency influences the propagation of α-synuclein pathology by promoting the release of extracellular vesicles and the cell-to-cell spread of α-synuclein aggregation. GBA1 knockdown in neuroblastoma cells increases the cell-to-cell transmission of α-synuclein aggregates, which is reversed by the ectopic expression of the wild-type GBA1 gene [150]. β-Glucocerebrosidase deficiency significantly increases the release of α-synuclein fibrils from differentiated human dopaminergic cells and promotes the transfer of α-synuclein pathology to naïve cells [151]. In p.L483P variant knock-in mice, greater formation and propagation of α-synuclein inclusions are observed throughout the brain after the injection of α-synuclein preformed fibrils into the striatum compared to the wild-type controls [152].
- Defective β-glucocerebrosidase activity increases the release of extracellular vesicles in fibroblasts derived from GBA1-PD [153]. In a mouse model of PD, pharmacological depletion of β- glucocerebrosidase or the overexpression of mutant β- glucocerebrosidase increases the secretion of exosome-associated and free-form oligomeric α-synuclein [154]. However, the levels of α-synuclein in plasma exosomes do not differ between GBA1-PD and idiopathic PD patients [155]. ER stress and autophagic-lysosomal perturbation are associated with elevated extracellular α-synuclein in dopaminergic neurons derived from the iPSCs of PD patients with the p.N409S variant [100].
- Mitochondrial dysfunction
- Mitochondrial impairment is well documented in the pathogenesis of PD via the complex interplay between oxidative stress and α-synuclein accumulation [156,157], as evidenced by several causative genes for PD, such as PINK1, PRKN, and PARK7, which are also involved in mitochondrial turnover. Hence, mitochondrial impairment may be an important mechanism to be considered for future therapeutic development for PD. A possible relationship among β-glucocerebrosidase deficiency, mitochondrial dysfunction, and α-synuclein aggregation has been suggested in experimental studies. A deficiency in β- glucocerebrosidase induced by conduritol B epoxide, shRNA, or GBA1 knockout causes mitochondrial fragmentation, reduces adenosine triphosphate synthesis, and increases free oxygen radicals in in vivo and in vitro models [74,158]. A link between β-glucocerebrosidase deficiency and mitochondrial dysfunction may be ascribed to the reduced autophagy-lysosomal clearance of damaged mitochondria, direct damage to mitochondria caused by accumulated α-synuclein, calcium dysregulation, and neuroinflammation due to the activation of glia through the accumulation of glucosylceramide [159].
- Modifier genes
- Low penetrance and phenotypic heterogeneity imply that other genetic or environmental factors may influence the expression of PD among individuals carrying a GBA1 variant. Genome-wide approaches and candidate gene studies have reported several modifier genes interacting with the GBA1-regulated pathway and modulating phenotypic expression. LIMP-2, encoded by SCARB2, has been suggested as a potential modifier gene [160,161]. LIMP-2 is a ubiquitously expressed transmembrane glycoprotein that transports β-glucocerebrosidase from the ER to the lysosomes through a mannose-6-phosphate receptor-independent pathway [160]. Several GWASs have demonstrated that SNPs, such as rs6812193 and rs6825004, within the SCARB2 gene are significantly associated with PD [16,161,162]. In the brains of LIMP2–deficient (LMIP-2-/-) mice, reduced β-glucocerebrosidase activity results in glucosylceramide storage, autophagy lysosomal defects, and α-synuclein accumulation, which is reversed with the overexpression of LIMP-2 [163]. In midbrain dopaminergic neurons of PD patients, LIMP-2 expression is increased, probably due to a compensatory mechanism for lysosomal β- glucocerebrosidase deficiency [163]. Another candidate modifier is the MTX1 gene located 10 kb downstream of GBA1, which encodes a mitochondrial protein. The MTX1 c.184T>A variant is more frequent in GBA1-PD patients, and the homozygous MTX1 c.184A/A genotype is significantly associated with a younger age at onset in GBA1-PD patients [164]. A GWAS identified BIN1, responsible for synaptic vesicle endocytosis in the CNS, as a modifier gene for GBA1-PD patients. rs13403026 in the BIN1 locus is associated with older age at onset in carriers of mild and severe GBA1 variants [165]. A recent eQTL analysis demonstrated that common noncoding SNPs within the GBA1 gene also regulate GBA1 expression and coregulate potential modifier genes, modulating the age at onset and motor and cognitive progression in GBA1-PD patients [166]. A recent GWAS revealed that rs356219 in the SNCA locus and rs1293298 in CTSB, encoding lysosomal cathepsin B, are significantly associated with the penetrance of GBA1 variants [167]. Furthermore, the SNCA rs356219 G/G polymorphism significantly accelerated the progression to HY 3 in GBA1-PD patients, suggesting a synergistic interaction between variants of the GBA1 and SNCA genes [168]. The findings of a recent in vitro study suggested a negative regulatory effect of the LRRK2 variant on lysosomal β-glucocerebrosidase activity by demonstrating that mutant leucine-rich repeat kinase decreases β- glucocerebrosidase activity in dopaminergic neurons derived from PD patients carrying a LRRK2 variant [169]. In this study, LRRK2 kinase inhibition increased β-glucocerebrosidase activity and reduced α-synuclein accumulation in dopaminergic neurons from PD patients with either LRRK2 or GBA1 variants. TMEM175, encoding a transmembrane lysosomal potassium channel, has also been proposed as a potential modifier gene. Two large-scale meta-analyses of PD GWAS have shown that the TMEM175/GAK/DGKQ locus is the third most significant peak [18,19]. Targeted sequencing of the coding and regulatory regions of TMEM175, GAK, and DGKQ, followed by the genotyping of 2 TMEM175 coding variants, found that TMEM175 p.M393T and p.Q65P were associated with PD. In addition, TMEM175 p.M393T was significantly associated with reduced β-glucocerebrosidase activity [170]. Functional studies of TMEM175 p.M393T demonstrated similar changes induced by TMEM175 knockout, but to a lesser degree, including reduced lysosomal expression of the TMEM175 protein, increased lysosomal pH, and reduced β- glucocerebrosidase activity [171]. In a recent study, the overall burden of deleterious variants for lysosomal storage disorders significantly increased the penetrance of GBA1 variants, mostly due to the presence of a second variant in GBA1 or variants in mucopolysaccharidosis-related genes [172].
PATHOPHYSIOLOGY LEADING TO TREATMENT AVENUES
- The strong relationship between GBA1 variants and PD has opened up the possibility that treatment approaches for GD can be applied as novel disease-modifying strategies for PD. Currently, there are two main treatment approaches for GD: 1) enhancing deficient β-glucocerebrosidase activity through enzyme replacement, gene therapy or SMCs and 2) reducing glucosylceramide substrate accumulation (Table 2). In addition to applying GBA1-targeted drugs, testing patients for their GBA1 variant status may help in the decision-making process for the current conventional therapies for PD, such as deep brain stimulation (DBS), due to the detrimental impact of GBA1 variant on treatment outcomes.
- Enzyme replacement therapy
- Enzyme replacement therapy (ERT) is a primary therapeutic option for GD. Periodic intravenous infusions of recombinant β-glucocerebrosidase effectively improve the visceral and hematological symptoms of GD patients and extend their life expectancy [173,174]. However, ERT is ineffective for the neurological symptoms of GD or PD patients [175]. This is because commercially available forms of ERTs are not able to cross the blood‒brain barrier (BBB) due to their size and the absence of mannose receptors on the brain endothelium [176]. Alternative approaches to enhance the delivery of β-glucocerebrosidase enzymes across the BBB are being examined.
- Ongoing trial. Magnetic resonance-guided focused ultrasound (MRgFUS) in combination with microbubbles can reversibly enhance BBB opening in the target brain region, enabling the intracerebral delivery of biological therapeutics with poor BBB permeability [177,178]. In an open-label phase I study, putaminal delivery of intravenous β-glucocerebrosidase by using microbubble-mediated MRgFUS showed favorable safety and feasibility in four PD patients carrying a GBA1 variant (NCT04370665) [179].
- Gene therapy
- To increase the levels of CNS recombinant β- glucocerebrosidase enzyme, direct intracerebral gene delivery with adeno-associated viral (AAV)-based vectors has been considered. A preclinical study showed that the restoration of β- glucocerebrosidase expression in the CNS via intracerebral injection of a GBA1-coding AAV vector reduces α-synuclein accumulation in A53T α- synuclein-overexpressing mice [94].
- Ongoing trial.Intraventricular treatment with PR001, an AAV9 vector containing GBA1, increases β-glucocerebrosidase expression, elevates β-glucocerebrosidase activity, reduces the accumulation of glycolipid substrates, and improves behavioral impairment in in vivo models of GBA1-PD [95]. A phase I/IIa randomized controlled trial is underway to assess the therapeutic efficacy of intracisternal administration of PR001 to GBA1-PD patients (ClinicalTrials.gov ID: NCT04127578).
- Substrate reduction therapy
- Given that the accumulation of glucosylceramide, a direct substrate of β-glucocerebrosidase, plays a key role in the pathogenesis of GD, reducing glucosylceramide by inhibiting glucosylceramide synthase could be an alternative therapeutic strategy for GD. Unfortunately, commercially available glucosylceramide synthase inhibitors, including miglustat and eliglustat, have limited efficacy for the neurological symptoms of GD patients due to their low BBB penetrance [180]. However, a novel BBB-penetrant glucosylceramide synthase inhibitor, venglustat, reduces the accumulation of glucosylceramide and α-synuclein and improves behavioral outcomes in the Gaucher-related or A53T-α-synuclein overexpressed mouse models [115].
- Completed trial. The results from these experimental studies led to a phase II clinical trial evaluating the effectiveness of venglustat for treating GBA1-PD patients (MOVES-PD, ClinicalTrials.gov identifier NCT02906020). In Part I of this trial, venglustat showed favorable safety and target engagement with a significant reduction in glucosylceramide in both CSF and plasma in a dose-dependent manner [118]. However, the results of part II of the study, reported at the MDS 2021 and AD/PD-2021 conferences, did not demonstrate that venglustat achieved significant clinical improvement [181]. Instead, GBA1-PD patients in the treatment arm showed a more rapid deterioration in motor and cognitive function, leading to the study’s premature discontinuation. Several possible hypotheses can explain the results of the MOVES-PD trial. Despite the clinical and experimental data, the accumulation of glucosylceramide may not be a major player in the pathogenesis of PD. A decreased level of glucosylceramide in neurons could disrupt sphingolipid metabolism, damaging the neuronal membrane. This is supported by the fact that peripheral neuropathy is reported as an adverse effect of SRT in patients with GD [182].
- Small molecule chaperones
- SMCs bind to an active or alternative site of an enzyme and aid in translocating it to the target organelle. SMCs are considered promising therapeutics for PD because they can bind to the β-glucocerebrosidase that is sequestered in the ER due to disrupted ER-Golgi trafficking and can facilitate its translocation to the lysosome [183]. This can enhance the enzymatic activity of β-glucocerebrosidase and relieve the overload of UPS caused by excessive ERADs. Another important advantage of SMCs is their ability to cross the BBB [184]. There are two types of SMCs: inhibitory SMCs that bind to an active site of the enzyme and noninhibitory SMCs that bind to an alternative site.
- Inhibitory SMCs directly interact with the active site of β- glucocerebrosidase and competitively inhibit the binding of glucosylceramide, which blocks β-glucocerebrosidase from hydrolyzing glucosylceramide. Inhibitory SMCs are detached from β-glucocerebrosidase in the lysosome where glucosylceramide is abundant so that β-glucocerebrosidase binds to and catalyzes glucosylceramide. Experimental studies have shown that the iminosugar isofagomine enhances β-glucocerebrosidase activity and reduces substrate accumulation in in vitro and in vivo models of GD [185-187]. However, in a clinical trial, type I GD patients treated with isofagomine did not show clinical improvement, probably due to its high binding affinity to β- glucocerebrosidase, and the advancement of the study was terminated (NCT00875160).
- Noninhibitory SMCs, second-generation pharmacological chaperones, are activators of β-glucocerebrosidase with chaperone activity. These BBB-penetrant small molecules bind to a region other than the active site, thereby minimizing the competitive inhibition with glucosylceramide at the active site of β- glucocerebrosidase [188]. Accordingly, noninhibitory SMCs enhance the translocation of β-glucocerebrosidase to lysosomes and activate the misfolded enzyme. A new quantitative high-throughput screening using spleen extracts from GD patients identified two series of compounds of β-glucocerebrosidase activators with chaperone activity, including a salicylic acid derivative and a pyrazolopyrimidine carboxamide derivative [189,190]. Treatment with NCGC607, a salicylic acid derivative analog of ML266, restores β-glucocerebrosidase activity and reduces the levels of glucosylceramide and glycosylsphingosine in both macrophages and dopaminergic neurons derived from the iPSCs of type I and type II GD patients [191]. Moreover, in dopaminergic neurons derived from the iPSCs of type II GD patients and GD patients with a PD phenotype, α-synuclein accumulation is significantly reversed by NGCC607, suggesting its potential efficacy for treating neuropathic GD and PD patients [191].
- A pyrazolopyrimidine derivative, NCGC758, also effectively increases β-glucocerebrosidase activity and reduces glycolipid storage in macrophages differentiated from monocytes or iPSCs of GD patients [192]. The therapeutic effect of NCGC758 was tested on iPSC-derived dopaminergic neurons from patients harboring PD-causing variants, including SNCA, GBA1, and PARK9, as well as idiopathic PD patients [193]. NCGC758 specifically enhances β-glucocerebrosidase activity in the lysosomal compartment and mitigates the levels of glucosylceramide and α-synuclein in these neuronal models of PD, regardless of variant status [193].
- Quinazoline derivatives have been identified as noniminosugar β-glucocerebrosidase inhibitors with chaperone activity through high throughput screening [194-196]. Quinazoline derivatives have been reported to be the most potent noniminosugar β-glucocerebrosidase inhibitors, selectively stabilizing β- glucocerebrosidase [196]. A recent structure-activity relationship study demonstrated that N-methylation transforms quinazoline compounds from inhibitors to activators of β-glucocerebrosidase [197]. The efficacy of S-181, an N-methylated quinazoline compound, was examined with iPSC-derived dopaminergic neurons from idiopathic PD patients and PD patients carrying GBA1, LRRK2, DJ-1, or PARKIN variants and PD mice with a heterozygous GBA1 variant [198]. Treatment with S-181 enhances β -glucocerebrosidase activity and lowers the accumulation of glucosylceramide and α-synuclein in both in vitro and in vivo PD models.
- Ongoing trial. LTI-291, another pyrazolopyrimidine, showed favorable safety and tolerability, without a significant change in glycolipid levels, in two phase I trials conducted on healthy middle-aged volunteers (Trialregister.nl ID: NL6421 and NL6516). LTI-291 is currently being tested for its therapeutic efficacy in GBA1-PD patients in a phase Ib randomized controlled trial (Trialregister.nl ID: NL6574).
- High-throughput screening using a thermal denaturation assay identified that ambroxol is a mixed-type inhibitor of β- glucocerebrosidase, with its inhibitory action being maximal at the neutral pH of the ER and being an activator of β- glucocerebrosidase at the acidic pH of lysosomes [199]. Ambroxol treatment significantly increases β-glucocerebrosidase activity in fibroblasts from GD patients [184] and PD patients carrying a heterozygous GBA1 variant [143]. In dopaminergic neurons derived from PD patients carrying the p.N409S variant, ambroxol treatment enhances β-glucocerebrosidase activity and reduces α-synuclein accumulation by restoring the autophagy lysosomal defect [144]. In the brains of mice expressing heterozygous p.L483P or mice overexpressing human α-synuclein, ambroxol treatment increases β-glucocerebrosidase activity and decreases α-synuclein [145]. High-dose oral administration of ambroxol increases β- glucocerebrosidase activity in the brains of wild-type nonhuman primates [146]. Moreover, an open-label pilot study in 5 patients with neuropathic GD showed that high-dose oral ambroxol in combination with ERT achieves a CSF ambroxol concentration that is 10%–20% of that of the serum ambroxol concentration, decreases the CSF glucosylsphingosine levels, and improves neurological manifestations [200]. This experimental and clinical evidence has led to two phase II clinical trials in PD patients.
- Completed trial. In a single-center, open-label noncontrolled clinical trial, 18 PD patients, including 8 with a GBA1 variant, were treated with 1.26 g/day ambroxol for six months (ClinicalTrials.gov identifier: NCT02941822). Ambroxol treatment increases the ambroxol level, decreases β-glucocerebrosidase activity, and increases α-synuclein levels in the CSF [147]. Ambroxol treatment may reduce β-glucocerebrosidase activity due to its inhibitory action on β-glucocerebrosidase activity in acellular human CSF with a neutral pH, while it will increase β-glucocerebrosidase activity in brain tissue with an acidic pH, as reported in animal studies [145,146]. Increased CSF α-synuclein levels after ambroxol treatment can be interpreted as an enhanced extracellular export of α-synuclein from the brain parenchyma. In addition, motor disability is improved after ambroxol treatment regardless of the presence of a GBA1 variant.
- Ongoing trial. A double-blind, randomized, placebo-controlled trial is underway to test whether ambroxol treatment can reduce the rate of cognitive decline in PD patients with dementia (ClinicalTrials.gov identifier: NCT02914366).
- GBA1-based decision-making for conventional therapies
- GBA1 variants are not only associated with a more severe clinical phenotype but also predict a more deleterious prognosis in terms of the response to conventional therapies. Accordingly, GBA1 variant status may provide a valuable clue for selecting therapeutic options to improve the treatment outcome. For instance, a worse DBS outcome has been reported in GBA1-PD patients, such as a lower reduction in levodopa equivalent daily dosage [201], more frequent nonmotor symptoms [202], and worse quality of life [202]. In addition, GBA1 variants are substantially associated with more profound cognitive decline after DBS [201-203], while improvements in motor disability, motor fluctuation, and dyskinesia are comparable between GBA1-PD and idiopathic PD patients [201-203]. Strikingly, a recent study demonstrated a combined effect of DBS and GBA1 variants on the longitudinal deterioration in cognition [204]. In this study, PD patients were categorized as GBA1 carriers with or without DBS and noncarriers with or without DBS, and the rate of change in cognition was compared among the groups over time. The authors reported that GBA1 carriers with DBS showed the most rapid cognitive decline among the various groups.
- However, studies investigating the association between GBA1 variant status and DBS outcome in PD patients are still limited and predominantly based on group-level data, not incorporating the large variability in an individual patient. In particular, youngonset PD patients who are professionally active and without cognitive impairment may still be good candidates for a particular treatment regardless of GBA1 variant status. Therefore, DBS candidates need to be carefully recommended and counseled regarding DBS surgery, and the GBA1 variant status and other clinical factors of an individual PD patient need to be considered.
THERAPEUTIC STRATEGIES TARGETING GBA1-REGULATED PATHWAYS
Inhibitory SMCs.
Noninhibitory SMCs.
Ambroxol.
- The recent advances in genetics and functional experiments related to GBA1 may provide essential information to develop future therapeutics for PD. GBA1 variants increase the risk for PD and may accelerate disease progression in PD patients, with more aggressive phenotypes observed in patients carrying a severe variant. Accordingly, future proof-of-concept trials need to stratify PD patients based on their GBA1 variant status (presence and genotype) to mitigate the innate bias. In clinical practice, testing PD patients for GBA1 variants may provide useful information to predict the outcomes of current conventional treatments, such as DBS, more accurately. Future therapeutic strategies targeting GBA1-regulated pathways could lead to disease modification for genetically at-risk individuals for PD. However, lessons should be learned from the recent failure of the MOVES-PD trial, in which the substrate-reducing agent effectively removed the glucosylceramide accumulation but failed to achieve clinical improvement. In addition, potential modifiers for the penetrance or expressivity of GBA1 variants and gene‒gene interactions may provide valuable insights into the development of novel neuroprotective agents for PD pathogenesis. In addition, the investigation of GBA1 variants in the non-European population may further increase the understanding of the implications of GBA1 variants in PD. Last, PD is a complex disease, hence recommendations and treatment decisions should be individualized, collectively considering and integrating GBA1 variant status, other genetic factors, and comprehensive clinical features.
CONCLUSIONS AND RECOMMENDATIONS
-
Conflicts of Interest
The authors have no financial conflicts of interest. Christine Klein is medical advisor to Centogene and Retromer Therapeutics and received Speakers’ honoraria from Desitin and Bial, however these do not affect the content of the manuscript.
-
Funding Statement
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea, funded by the Ministry of Science and ICT (2021R1C1C1010753). This study was also supported by the Asan Institute for Life Sciences, Asan Medical Center, Seoul, Republic of Korea (grant number: 2022IT0012).
-
Author Contributions
Conceptualization: Young Eun Huh, Christine Klein, Sun Ju Chung. Funding acquisition: Young Eun Huh. Supervision: Christine Klein, Sun Ju Chung. Visualization: Young Eun Huh, Clemens R. Scherzer. Writing—original draft: Young Eun Huh. Writing—review & editing: all authors.
Notes
PD, Parkinson’s disease; GBA1, glucosylceramidase beta 1; GBA1-PD, Parkinson’s disease patients with a GBA1 mutation; idiopathic PD, Parkinson’s disease patients without GBA1 mutation; HR, hazard ratio; ns, not significant; HY, Hoehn and Yahr; REM, rapid eye movement; DBS, deep brain stimulation; LEDD, levodopa equivalent daily dosage; QOL, quality of life.
- 1. Zhang PL, Chen Y, Zhang CH, Wang YX, Fernandez-Funez P. Genetics of Parkinson’s disease and related disorders. J Med Genet 2018;55:73–80.ArticlePubMed
- 2. Lubbe S, Morris HR. Recent advances in Parkinson’s disease genetics. J Neurol 2014;261:259–266.ArticlePubMedPDF
- 3. Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol 2019;18:1091–1102.PubMedPMC
- 4. Brady RO, Kanfer JN, Bradley RM, Shapiro D. Demonstration of a deficiency of glucocerebroside-cleaving enzyme in Gaucher’s disease. J Clin Invest 1966;45:1112–1115.ArticlePubMedPMC
- 5. Jmoudiak M, Futerman AH. Gaucher disease: pathological mechanisms and modern management. Br J Haematol 2005;129:178–188.ArticlePubMed
- 6. Grabowski GA. Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet 2008;372:1263–1271.ArticlePubMed
- 7. Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 2009;361:1651–1661.ArticlePubMedPMC
- 8. Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat 2008;29:567–583.ArticlePubMed
- 9. Neudorfer O, Giladi N, Elstein D, Abrahamov A, Turezkite T, Aghai E, et al. Occurrence of Parkinson’s syndrome in type I Gaucher disease. QJM 1996;89:691–694.ArticlePubMed
- 10. Halperin A, Elstein D, Zimran A. Increased incidence of Parkinson disease among relatives of patients with Gaucher disease. Blood Cells Mol Dis 2006;36:426–428.ArticlePubMed
- 11. Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 2009;41:1308–1312.ArticlePubMedPMCPDF
- 12. Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet 2009;41:1303–1307.ArticlePubMedPDF
- 13. Pankratz N, Wilk JB, Latourelle JC, DeStefano AL, Halter C, Pugh EW, et al. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet 2009;124:593–605.ArticlePubMedPMCPDF
- 14. Sidransky E, Lopez G. The link between the GBA gene and parkinsonism. Lancet Neurol 2012;11:986–998.ArticlePubMedPMC
- 15. International Parkinson Disease Genomics Consortium. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet 2011;377:641–649.ArticlePubMedPMC
- 16. Do CB, Tung JY, Dorfman E, Kiefer AK, Drabant EM, Francke U, et al. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet 2011;7:e1002141. ArticlePubMedPMC
- 17. Pankratz N, Beecham GW, DeStefano AL, Dawson TM, Doheny KF, Factor SA, et al. Meta-analysis of Parkinson’s disease: identification of a novel locus, RIT2. Ann Neurol 2012;71:370–384.ArticlePubMedPMC
- 18. Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 2014;46:989–993.ArticlePubMedPMCPDF
- 19. Chang D, Nalls MA, Hallgrímsdóttir IB, Hunkapiller J, van der Brug M, Cai F, et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 2017;49:1511–1516.ArticlePubMedPMCPDF
- 20. Saunders-Pullman R, Hagenah J, Dhawan V, Stanley K, Pastores G, Sathe S, et al. Gaucher disease ascertained through a Parkinson’s center: imaging and clinical characterization. Mov Disord 2010;25:1364–1372.ArticlePubMedPMC
- 21. Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R. Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N Engl J Med 2004;351:1972–1977.ArticlePubMed
- 22. Migdalska-Richards A, Schapira AH. The relationship between glucocerebrosidase mutations and Parkinson disease. J Neurochem 2016;139 Suppl 1:77–90.ArticlePubMedPMC
- 23. De Marco EV, Annesi G, Tarantino P, Rocca FE, Provenzano G, Civitelli D, et al. Glucocerebrosidase gene mutations are associated with Parkinson’s disease in southern Italy. Mov Disord 2008;23:460–463.ArticlePubMed
- 24. Kalinderi K, Bostantjopoulou S, Paisan-Ruiz C, Katsarou Z, Hardy J, Fidani L. Complete screening for glucocerebrosidase mutations in Parkinson disease patients from Greece. Neurosci Lett 2009;452:87–89.ArticlePubMed
- 25. Liu G, Boot B, Locascio JJ, Jansen IE, Winder-Rhodes S, Eberly S, et al. Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann Neurol 2016;80:674–685.ArticlePubMedPMCPDF
- 26. Tan EK, Tong J, Fook-Chong S, Yih Y, Wong MC, Pavanni R, et al. Glucocerebrosidase mutations and risk of Parkinson disease in Chinese patients. Arch Neurol 2007;64:1056–1058.ArticlePubMed
- 27. Sun QY, Guo JF, Wang L, Yu RH, Zuo X, Yao LY, et al. Glucocerebrosidase gene L444P mutation is a risk factor for Parkinson’s disease in Chinese population. Mov Disord 2010;25:1005–1011.ArticlePubMed
- 28. Wu YR, Chen CM, Chao CY, Ro LS, Lyu RK, Chang KH, et al. Glucocerebrosidase gene mutation is a risk factor for early onset of Parkinson disease among Taiwanese. J Neurol Neurosurg Psychiatry 2007;78:977–979.ArticlePubMedPMC
- 29. Choi JM, Kim WC, Lyoo CH, Kang SY, Lee PH, Baik JS, et al. Association of mutations in the glucocerebrosidase gene with Parkinson disease in a Korean population. Neurosci Lett 2012;514:12–15.ArticlePubMed
- 30. Mitsui J, Mizuta I, Toyoda A, Ashida R, Takahashi Y, Goto J, et al. Mutations for Gaucher disease confer high susceptibility to Parkinson disease. Arch Neurol 2009;66:571–576.ArticlePubMed
- 31. Goker-Alpan O, Schiffmann R, LaMarca ME, Nussbaum RL, McInerney-Leo A, Sidransky E. Parkinsonism among Gaucher disease carriers. J Med Genet 2004;41:937–940.ArticlePubMedPMC
- 32. Straniero L, Asselta R, Bonvegna S, Rimoldi V, Melistaccio G, Soldà G, et al. The SPID-GBA study: sex distribution, penetrance, incidence, and dementia in GBA-PD. Neurol Genet 2020;6:e523. ArticlePubMedPMC
- 33. Sidransky E. Gaucher disease and parkinsonism. Mol Genet Metab 2005;84:302–304.ArticlePubMed
- 34. Rana HQ, Balwani M, Bier L, Alcalay RN. Age-specific Parkinson disease risk in GBA mutation carriers: information for genetic counseling. Genet Med 2013;15:146–149.ArticlePubMedPMCPDF
- 35. Alcalay RN, Dinur T, Quinn T, Sakanaka K, Levy O, Waters C, et al. Comparison of Parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol 2014;71:752–757.ArticlePubMedPMC
- 36. Höglinger G, Schulte C, Jost WH, Storch A, Woitalla D, Krüger R, et al. GBA-associated PD: chances and obstacles for targeted treatment strategies. J Neural Transm (Vienna) 2022;129:1219–1233.ArticlePubMedPMCPDF
- 37. Mata IF, Leverenz JB, Weintraub D, Trojanowski JQ, Chen-Plotkin A, Van Deerlin VM, et al. GBA variants are associated with a distinct pattern of cognitive deficits in Parkinson’s disease. Mov Disord 2016;31:95–102.ArticlePubMedPMCPDF
- 38. Nichols WC, Pankratz N, Marek DK, Pauciulo MW, Elsaesser VE, Halter CA, et al. Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology 2009;72:310–316.ArticlePubMedPMC
- 39. Davis MY, Johnson CO, Leverenz JB, Weintraub D, Trojanowski JQ, Chen-Plotkin A, et al. Association of GBA mutations and the E326K polymorphism with motor and cognitive progression in Parkinson disease. JAMA Neurol 2016;73:1217–1224.ArticlePubMedPMC
- 40. Stoker TB, Camacho M, Winder-Rhodes S, Liu G, Scherzer CR, Foltynie T, et al. Impact of GBA1 variants on long-term clinical progression and mortality in incident Parkinson’s disease. J Neurol Neurosurg Psychiatry 2020;91:695–702.ArticlePubMedPMC
- 41. Iwaki H, Blauwendraat C, Leonard HL, Liu G, Maple-Grødem J, Corvol JC, et al. Genetic risk of Parkinson disease and progression: an analysis of 13 longitudinal cohorts. Neurol Genet 2019;5:e348. ArticlePubMedPMC
- 42. Neumann J, Bras J, Deas E, O’Sullivan SS, Parkkinen L, Lachmann RH, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 2009;132:1783–1794.ArticlePubMedPMC
- 43. Parkkinen L, Neumann J, O’Sullivan SS, Holton JL, Revesz T, Hardy J, et al. Glucocerebrosidase mutations do not cause increased Lewy body pathology in Parkinson’s disease. Mol Genet Metab 2011;103:410–412.ArticlePubMed
- 44. Nalls MA, Duran R, Lopez G, Kurzawa-Akanbi M, McKeith IG, Chinnery PF, et al. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol 2013;70:727–735.ArticlePubMedPMC
- 45. Gan-Or Z, Amshalom I, Kilarski LL, Bar-Shira A, Gana-Weisz M, Mirelman A, et al. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 2015;84:880–887.ArticlePubMedPMC
- 46. Cilia R, Tunesi S, Marotta G, Cereda E, Siri C, Tesei S, et al. Survival and dementia in GBA-associated Parkinson’s disease: the mutation matters. Ann Neurol 2016;80:662–673.PubMed
- 47. Zhang Y, Sun QY, Zhao YW, Shu L, Guo JF, Xu Q, et al. Effect of GBA mutations on phenotype of Parkinson’s disease: a study on Chinese population and a meta-analysis. Parkinsons Dis 2015;2015:916971.ArticlePubMedPMCPDF
- 48. Clark LN, Ross BM, Wang Y, Mejia-Santana H, Harris J, Louis ED, et al. Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology 2007;69:1270–1277.ArticlePubMedPMC
- 49. Jesús S, Huertas I, Bernal-Bernal I, Bonilla-Toribio M, Cáceres-Redondo MT, Vargas-González L, et al. GBA variants influence motor and nonmotor features of Parkinson’s disease. PLoS One 2016;11:e0167749. ArticlePubMedPMC
- 50. Mata IF, Samii A, Schneer SH, Roberts JW, Griffith A, Leis BC, et al. Glucocerebrosidase gene mutations: a risk factor for Lewy body disorders. Arch Neurol 2008;65:379–382.PubMedPMC
- 51. Gan-Or Z, Giladi N, Rozovski U, Shifrin C, Rosner S, Gurevich T, et al. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 2008;70:2277–2283.ArticlePubMed
- 52. Malek N, Weil RS, Bresner C, Lawton MA, Grosset KA, Tan M, et al. Features of GBA-associated Parkinson’s disease at presentation in the UK Tracking Parkinson’s study. J Neurol Neurosurg Psychiatry 2018;89:702–709.ArticlePubMedPMC
- 53. Thaler A, Gurevich T, Bar Shira A, Gana Weisz M, Ash E, Shiner T, et al. A “dose” effect of mutations in the GBA gene on Parkinson’s disease phenotype. Parkinsonism Relat Disord 2017;36:47–51.ArticlePubMed
- 54. Winder-Rhodes SE, Evans JR, Ban M, Mason SL, Williams-Gray CH, Foltynie T, et al. Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain 2013;136:392–399.ArticlePubMed
- 55. Lopez G, Kim J, Wiggs E, Cintron D, Groden C, Tayebi N, et al. Clinical course and prognosis in patients with Gaucher disease and parkinsonism. Neurol Genet 2016;2:e57. ArticlePubMedPMC
- 56. Rossi M, Castillo-Torres SA, Merello M. Early motor response to dopamine replacement therapy in Parkinson’s disease patients carrying GBA variants. J Neurol Sci 2022;440:120354.ArticlePubMed
- 57. Creese B, Bell E, Johar I, Francis P, Ballard C, Aarsland D. Glucocerebrosidase mutations and neuropsychiatric phenotypes in Parkinson’s disease and Lewy body dementias: review and meta-analyses. Am J Med Genet B Neuropsychiatr Genet 2018;177:232–241.ArticlePubMedPDF
- 58. Brockmann K, Srulijes K, Pflederer S, Hauser AK, Schulte C, Maetzler W, et al. GBA-associated Parkinson’s disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov Disord 2015;30:407–411.ArticlePubMedPDF
- 59. Oeda T, Umemura A, Mori Y, Tomita S, Kohsaka M, Park K, et al. Impact of glucocerebrosidase mutations on motor and nonmotor complications in Parkinson’s disease. Neurobiol Aging 2015;36:3306–3313.ArticlePubMed
- 60. Brockmann K, Srulijes K, Hauser AK, Schulte C, Csoti I, Gasser T, et al. GBA-associated PD presents with nonmotor characteristics. Neurology 2011;77:276–280.ArticlePubMed
- 61. Swan M, Doan N, Ortega RA, Barrett M, Nichols W, Ozelius L, et al. Neuropsychiatric characteristics of GBA-associated Parkinson disease. J Neurol Sci 2016;370:63–69.ArticlePubMedPMC
- 62. Beavan M, McNeill A, Proukakis C, Hughes DA, Mehta A, Schapira AH. Evolution of prodromal clinical markers of Parkinson disease in a GBA mutation-positive cohort. JAMA Neurol 2015;72:201–208.ArticlePubMedPMC
- 63. Gan-Or Z, Mirelman A, Postuma RB, Arnulf I, Bar-Shira A, Dauvilliers Y, et al. GBA mutations are associated with rapid eye movement sleep behavior disorder. Ann Clin Transl Neurol 2015;2:941–945.ArticlePubMedPMCPDF
- 64. Gámez-Valero A, Iranzo A, Serradell M, Vilas D, Santamaria J, Gaig C, et al. Glucocerebrosidase gene variants are accumulated in idiopathic REM sleep behavior disorder. Parkinsonism Relat Disord 2018;50:94–98.ArticlePubMed
- 65. Krohn L, Ruskey JA, Rudakou U, Leveille E, Asayesh F, Hu MTM, et al. GBA variants in REM sleep behavior disorder: a multicenter study. Neurology 2020;95:e1008–e1016.ArticlePubMedPMC
- 66. Goker-Alpan O, Masdeu JC, Kohn PD, Ianni A, Lopez G, Groden C, et al. The neurobiology of glucocerebrosidase-associated parkinsonism: a positron emission tomography study of dopamine synthesis and regional cerebral blood flow. Brain 2012;135:2440–2448.ArticlePubMedPMC
- 67. Walker Z, Costa DC, Walker RW, Lee L, Livingston G, Jaros E, et al. Striatal dopamine transporter in dementia with Lewy bodies and Parkinson disease: a comparison. Neurology 2004;62:1568–1572.ArticlePubMed
- 68. McNeill A, Wu RM, Tzen KY, Aguiar PC, Arbelo JM, Barone P, et al. Dopaminergic neuronal imaging in genetic Parkinson’s disease: insights into pathogenesis. PLoS One 2013;8:e69190. ArticlePubMedPMC
- 69. Caminiti SP, Carli G, Avenali M, Blandini F, Perani D. Clinical and dopamine transporter imaging trajectories in a cohort of Parkinson’s disease patients with GBA mutations. Mov Disord 2022;37:106–118.PubMed
- 70. Simuni T, Brumm MC, Uribe L, Caspell-Garcia C, Coffey CS, Siderowf A, et al. Clinical and dopamine transporter imaging characteristics of leucine rich repeat kinase 2 (LRRK2) and glucosylceramidase beta (GBA) Parkinson’s disease participants in the Parkinson’s progression markers initiative: a cross-sectional study. Mov Disord 2020;35:833–844.ArticlePubMedPMCPDF
- 71. Chung SJ, Lee PH, Sohn YH, Kim YJ. Glucocerebrosidase mutations and motor reserve in Parkinson’s disease. J Parkinsons Dis 2021;11:1715–1724.ArticlePubMed
- 72. Arkadir D, Dinur T, Mullin S, Mehta A, Baris HN, Alcalay RN, et al. Trio approach reveals higher risk of PD in carriers of severe vs. mild GBA mutations. Blood Cells Mol Dis 2018;68:115–116.ArticlePubMed
- 73. Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, et al. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011;146:37–52.ArticlePubMedPMC
- 74. Cleeter MW, Chau KY, Gluck C, Mehta A, Hughes DA, Duchen M, et al. Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem Int 2013;62:1–7.ArticlePubMedPMC
- 75. Manning-Boğ AB, Schüle B, Langston JW. Alpha-synuclein-glucocerebrosidase interactions in pharmacological Gaucher models: a biological link between Gaucher disease and parkinsonism. Neurotoxicology 2009;30:1127–1132.ArticlePubMed
- 76. Xu YH, Sun Y, Ran H, Quinn B, Witte D, Grabowski GA. Accumulation and distribution of α-synuclein and ubiquitin in the CNS of Gaucher disease mouse models. Mol Genet Metab 2011;102:436–447.ArticlePubMedPMC
- 77. Schondorf DC, Aureli M, McAllister FE, Hindley CJ, Mayer F, Schmid B, et al. iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat Commun 2014;5:4028.ArticlePubMedPDF
- 78. Cullen V, Sardi SP, Ng J, Xu YH, Sun Y, Tomlinson JJ, et al. Acid β- glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter α-synuclein processing. Ann Neurol 2011;69:940–953.ArticlePubMed
- 79. Gegg ME, Sweet L, Wang BH, Shihabuddin LS, Sardi SP, Schapira AH. No evidence for substrate accumulation in Parkinson brains with GBA mutations. Mov Disord 2015;30:1085–1089.ArticlePubMedPMC
- 80. Gegg ME, Burke D, Heales SJ, Cooper JM, Hardy J, Wood NW, et al. Glucocerebrosidase deficiency in substantia nigra of Parkinson disease brains. Ann Neurol 2012;72:455–463.ArticlePubMedPMCPDF
- 81. Moors TE, Paciotti S, Ingrassia A, Quadri M, Breedveld G, Tasegian A, et al. Characterization of brain lysosomal activities in GBA-related and sporadic Parkinson’s disease and dementia with Lewy bodies. Mol Neurobiol 2019;56:1344–1355.ArticlePubMedPMCPDF
- 82. Parnetti L, Paciotti S, Eusebi P, Dardis A, Zampieri S, Chiasserini D, et al. Cerebrospinal fluid β-glucocerebrosidase activity is reduced in Parkinson’s disease patients. Mov Disord 2017;32:1423–1431.ArticlePubMedPDF
- 83. Parnetti L, Chiasserini D, Persichetti E, Eusebi P, Varghese S, Qureshi MM, et al. Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson’s disease. Mov Disord 2014;29:1019–1027.ArticlePubMedPMCPDF
- 84. Alcalay RN, Wolf P, Chiang MSR, Helesicova K, Zhang XK, Merchant K, et al. Longitudinal measurements of glucocerebrosidase activity in Parkinson’s patients. Ann Clin Transl Neurol 2020;7:1816–1830.ArticlePubMedPMCPDF
- 85. Huh YE, Chiang MSR, Locascio JJ, Liao Z, Liu G, Choudhury K, et al. β-glucocerebrosidase activity in GBA-linked Parkinson disease: the type of mutation matters. Neurology 2020;95:e685–e696.ArticlePubMedPMC
- 86. Pchelina S, Emelyanov A, Baydakova G, Andoskin P, Senkevich K, Nikolaev M, et al. Oligomeric α-synuclein and glucocerebrosidase activity levels in GBA-associated Parkinson’s disease. Neurosci Lett 2017;636:70–76.ArticlePubMed
- 87. Alcalay RN, Levy OA, Waters CC, Fahn S, Ford B, Kuo SH, et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain 2015;138:2648–2658.ArticlePubMedPMC
- 88. Papagiannakis N, Xilouri M, Koros C, Stamelou M, Antonelou R, Maniati M, et al. Lysosomal alterations in peripheral blood mononuclear cells of Parkinson’s disease patients. Mov Disord 2015;30:1830–1834.ArticlePubMed
- 89. Atashrazm F, Hammond D, Perera G, Dobson-Stone C, Mueller N, Pickford R, et al. Reduced glucocerebrosidase activity in monocytes from patients with Parkinson’s disease. Sci Rep 2018;8:15446.ArticlePubMedPMCPDF
- 90. Chiasserini D, Paciotti S, Eusebi P, Persichetti E, Tasegian A, KurzawaAkanbi M, et al. Selective loss of glucocerebrosidase activity in sporadic Parkinson’s disease and dementia with Lewy bodies. Mol Neurodegener 2015;10:15.ArticlePubMedPMCPDF
- 91. Murphy KE, Gysbers AM, Abbott SK, Tayebi N, Kim WS, Sidransky E, et al. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain 2014;137:834–848.ArticlePubMedPMC
- 92. Balducci C, Pierguidi L, Persichetti E, Parnetti L, Sbaragli M, Tassi C, et al. Lysosomal hydrolases in cerebrospinal fluid from subjects with Parkinson’s disease. Mov Disord 2007;22:1481–1484.ArticlePubMedPDF
- 93. Kim HJ, Jeon B, Song J, Lee WW, Park H, Shin CW. Leukocyte glucocerebrosidase and β-hexosaminidase activity in sporadic and genetic Parkinson disease. Parkinsonism Relat Disord 2016;23:99–101.ArticlePubMed
- 94. Sardi SP, Clarke J, Viel C, Chan M, Tamsett TJ, Treleaven CM, et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc Natl Acad Sci U S A 2013;110:3537–3542.ArticlePubMedPMC
- 95. Abeliovich A, Hefti F, Sevigny J. Gene therapy for Parkinson’s disease associated with GBA1 mutations. J Parkinsons Dis 2021;11(s2):S183–S188.ArticlePubMedPMC
- 96. Sardi SP, Clarke J, Kinnecom C, Tamsett TJ, Li L, Stanek LM, et al. CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc Natl Acad Sci U S A 2011;108:12101–12106.PubMedPMC
- 97. Gan-Or Z, Dion PA, Rouleau GA. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy 2015;11:1443–1457.ArticlePubMedPMC
- 98. Alcalay RN, Wolf P, Levy OA, Kang UJ, Waters C, Fahn S, et al. Alpha galactosidase A activity in Parkinson’s disease. Neurobiol Dis 2018;112:85–90.ArticlePubMedPMC
- 99. Wu G, Yan B, Wang X, Feng X, Zhang A, Xu X, et al. Decreased activities of lysosomal acid alpha-D-galactosidase A in the leukocytes of sporadic Parkinson’s disease. J Neurol Sci 2008;271:168–173.ArticlePubMed
- 100. Fernandes HJ, Hartfield EM, Christian HC, Emmanoulidou E, Zheng Y, Booth H, et al. ER stress and autophagic perturbations lead to elevated extracellular α-synuclein in GBA-N370S Parkinson’s iPSC-derived dopamine neurons. Stem Cell Reports 2016;6:342–356.ArticlePubMedPMC
- 101. Du TT, Wang L, Duan CL, Lu LL, Zhang JL, Gao G, et al. GBA deficiency promotes SNCA/alpha-synuclein accumulation through autophagic inhibition by inactivated PPP2A. Autophagy 2015;11:1803–1820.ArticlePubMedPMC
- 102. Magalhaes J, Gegg ME, Migdalska-Richards A, Doherty MK, Whitfield PD, Schapira AH. Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: relevance to Parkinson disease. Hum Mol Genet 2016;25:3432–3445.ArticlePubMedPMC
- 103. Kuo SH, Tasset I, Cheng MM, Diaz A, Pan MK, Lieberman OJ, et al. Mutant glucocerebrosidase impairs alpha-synuclein degradation by blockade of chaperone-mediated autophagy. Sci Adv 2022;8:eabm6393.PubMed
- 104. Zimmer KP, le Coutre P, Aerts HM, Harzer K, Fukuda M, O’Brien JS, et al. Intracellular transport of acid beta-glucosidase and lysosome-associated membrane proteins is affected in Gaucher’s disease (G202R mutation). J Pathol 1999;188:407–414.ArticlePubMed
- 105. Bonifacino JS, Weissman AM. Ubiquitin and the control of protein fate in the secretory and endocytic pathways. Annu Rev Cell Dev Biol 1998;14:19–57.ArticlePubMedPMC
- 106. Brodsky JL, McCracken AA. ER protein quality control and proteasomemediated protein degradation. Semin Cell Dev Biol 1999;10:507–513.ArticlePubMed
- 107. Bendikov-Bar I, Horowitz M. Gaucher disease paradigm: from ERAD to comorbidity. Hum Mutat 2012;33:1398–1407.ArticlePubMed
- 108. Maor G, Rencus-Lazar S, Filocamo M, Steller H, Segal D, Horowitz M. Unfolded protein response in Gaucher disease: from human to Drosophila. Orphanet J Rare Dis 2013;8:140.ArticlePubMedPMCPDF
- 109. Ron I, Horowitz M. ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum Mol Genet 2005;14:2387–2398.ArticlePubMed
- 110. Enquist IB, Lo Bianco C, Ooka A, Nilsson E, Mansson JE, Ehinger M, et al. Murine models of acute neuronopathic Gaucher disease. Proc Natl Acad Sci U S A 2007;104:17483–17488.ArticlePubMedPMC
- 111. Farfel-Becker T, Vitner E, Dekel H, Leshem N, Enquist IB, Karlsson S, et al. No evidence for activation of the unfolded protein response in neuronopathic models of Gaucher disease. Hum Mol Genet 2009;18:1482–1488.ArticlePubMed
- 112. Suzuki M, Fujikake N, Takeuchi T, Kohyama-Koganeya A, Nakajima K, Hirabayashi Y, et al. Glucocerebrosidase deficiency accelerates the accumulation of proteinase K-resistant α-synuclein and aggravates neurodegeneration in a Drosophila model of Parkinson’s disease. Hum Mol Genet 2015;24:6675–6686.ArticlePubMed
- 113. Zunke F, Moise AC, Belur NR, Gelyana E, Stojkovska I, Dzaferbegovic H, et al. Reversible conformational conversion of α-Synuclein into toxic assemblies by glucosylceramide. Neuron 2018;97:92–107.e10.ArticlePubMedPMC
- 114. Kim S, Yun SP, Lee S, Umanah GE, Bandaru VVR, Yin X, et al. GBA1 deficiency negatively affects physiological alpha-synuclein tetramers and related multimers. Proc Natl Acad Sci U S A 2018;115:798–803.PubMedPMC
- 115. Sardi SP, Viel C, Clarke J, Treleaven CM, Richards AM, Park H, et al. Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc Natl Acad Sci U S A 2017;114:2699–2704.ArticlePubMedPMC
- 116. Guedes LC, Chan RB, Gomes MA, Conceição VA, Machado RB, Soares T, et al. Serum lipid alterations in GBA-associated Parkinson’s disease. Parkinsonism Relat Disord 2017;44:58–65.ArticlePubMed
- 117. Huh YE, Park H, Chiang MSR, Tuncali I, Liu G, Locascio JJ, et al. Glucosylceramide in cerebrospinal fluid of patients with GBA-associated and idiopathic Parkinson’s disease enrolled in PPMI. NPJ Parkinsons Dis 2021;7:102.ArticlePubMedPMCPDF
- 118. Peterschmitt MJ, Saiki H, Hatano T, Gasser T, Isaacson SH, Gaemers SJM, et al. Safety, pharmacokinetics, and pharmacodynamics of oral Venglustat in patients with Parkinson’s disease and a GBA mutation: results from Part 1 of the randomized, double-blinded, placebo-controlled MOVES-PD trial. J Parkinsons Dis 2022;12:557–570.ArticlePubMedPMC
- 119. Taguchi YV, Liu J, Ruan J, Pacheco J, Zhang X, Abbasi J, et al. Glucosylsphingosine promotes α-synuclein pathology in mutant GBA-associated Parkinson’s disease. J Neurosci 2017;37:9617–9631.ArticlePubMedPMC
- 120. Ikuno M, Yamakado H, Akiyama H, Parajuli LK, Taguchi K, Hara J, et al. GBA haploinsufficiency accelerates alpha-synuclein pathology with altered lipid metabolism in a prodromal model of Parkinson’s disease. Hum Mol Genet 2019;28:1894–1904.ArticlePubMed
- 121. Heinrich M, Wickel M, Schneider-Brachert W, Sandberg C, Gahr J, Schwandner R, et al. Cathepsin D targeted by acid sphingomyelinasederived ceramide. EMBO J 1999;18:5252–5263.ArticlePubMedPMC
- 122. Liu F, Li X, Lu C, Bai A, Bielawski J, Bielawska A, et al. Ceramide activates lysosomal cathepsin B and cathepsin D to attenuate autophagy and induces ER stress to suppress myeloid-derived suppressor cells. Oncotarget 2016;7:83907–83925.ArticlePubMedPMC
- 123. McGlinchey RP, Lee JC. Cysteine cathepsins are essential in lysosomal degradation of α-synuclein. Proc Natl Acad Sci U S A 2015;112:9322–9327.ArticlePubMedPMC
- 124. Sevlever D, Jiang P, Yen SH. Cathepsin D is the main lysosomal enzyme involved in the degradation of alpha-synuclein and generation of its carboxy-terminally truncated species. Biochemistry 2008;47:9678–9687.ArticlePubMed
- 125. Qiao L, Hamamichi S, Caldwell KA, Caldwell GA, Yacoubian TA, Wilson S, et al. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol Brain 2008;1:17.ArticlePubMedPMCPDF
- 126. Cullen V, Lindfors M, Ng J, Paetau A, Swinton E, Kolodziej P, et al. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol Brain 2009;2:5.ArticlePubMedPMCPDF
- 127. Stoka V, Turk V, Turk B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res Rev 2016;32:22–37.ArticlePubMed
- 128. Kim MJ, Jeon S, Burbulla LF, Krainc D. Acid ceramidase inhibition ameliorates α-synuclein accumulation upon loss of GBA1 function. Hum Mol Genet 2018;27:1972–1988.ArticlePubMedPMC
- 129. Gan-Or Z, Liong C, Alcalay RN. GBA-associated Parkinson’s disease and other synucleinopathies. Curr Neurol Neurosci Rep 2018;18:44.ArticlePubMedPDF
- 130. Clark LN, Chan R, Cheng R, Liu X, Park N, Parmalee N, et al. Genewise association of variants in four lysosomal storage disorder genes in neuropathologically confirmed Lewy body disease. PLoS One 2015;10:e0125204. ArticlePubMedPMC
- 131. Dagan E, Schlesinger I, Ayoub M, Mory A, Nassar M, Kurolap A, et al. The contribution of Niemann-Pick SMPD1 mutations to Parkinson disease in Ashkenazi Jews. Parkinsonism Relat Disord 2015;21:1067–1071.ArticlePubMed
- 132. Gan-Or Z, Ozelius LJ, Bar-Shira A, Saunders-Pullman R, Mirelman A, Kornreich R, et al. The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology 2013;80:1606–1610.ArticlePubMedPMC
- 133. Abbott SK, Li H, Muñoz SS, Knoch B, Batterham M, Murphy KE, et al. Altered ceramide acyl chain length and ceramide synthase gene expression in Parkinson’s disease. Mov Disord 2014;29:518–526.ArticlePubMedPDF
- 134. Lerche S, Schulte C, Wurster I, Machetanz G, Roeben B, Zimmermann M, et al. The mutation matters: CSF profiles of GCase, sphingolipids, α-synuclein in PD(GBA). Mov Disord 2021;36:1216–1228.ArticlePubMedPDF
- 135. Mielke MM, Maetzler W, Haughey NJ, Bandaru VV, Savica R, Deuschle C, et al. Plasma ceramide and glucosylceramide metabolism is altered in sporadic Parkinson’s disease and associated with cognitive impairment: a pilot study. PLoS One 2013;8:e73094. ArticlePubMedPMC
- 136. Dettmer U, Newman AJ, Soldner F, Luth ES, Kim NC, von Saucken VE, et al. Parkinson-causing α-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat Commun 2015;6:7314.ArticlePubMedPMCPDF
- 137. Glajch KE, Moors TE, Chen Y, Bechade PA, Nam AY, Rajsombath MM, et al. Wild-type GBA1 increases the α-synuclein tetramer-monomer ratio, reduces lipid-rich aggregates, and attenuates motor and cognitive deficits in mice. Proc Natl Acad Sci U S A 2021;118:e2103425118. ArticlePubMedPMC
- 138. Galvagnion C, Marlet FR, Cerri S, Schapira AHV, Blandini F, Di Monte DA. Sphingolipid changes in Parkinson L444P GBA mutation fibroblasts promote α-synuclein aggregation. Brain 2022;145:1038–1051.ArticlePubMedPMCPDF
- 139. Gonzalez A, Valeiras M, Sidransky E, Tayebi N. Lysosomal integral membrane protein-2: a new player in lysosome-related pathology. Mol Genet Metab 2014;111:84–91.ArticlePubMedPMC
- 140. Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006;313:324–328.ArticlePubMedPMC
- 141. Chung CY, Khurana V, Auluck PK, Tardiff DF, Mazzulli JR, Soldner F, et al. Identification and rescue of α-synuclein toxicity in Parkinson patient-derived neurons. Science 2013;342:983–987.ArticlePubMedPMC
- 142. Mazzulli JR, Zunke F, Isacson O, Studer L, Krainc D. α-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc Natl Acad Sci U S A 2016;113:1931–1936.ArticlePubMedPMC
- 143. McNeill A, Magalhaes J, Shen C, Chau KY, Hughes D, Mehta A, et al. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain 2014;137:1481–1495.ArticlePubMedPMC
- 144. Yang SY, Beavan M, Chau KY, Taanman JW, Schapira AHV. A human neural crest stem cell-derived dopaminergic neuronal model recapitulates biochemical abnormalities in GBA1 mutation carriers. Stem Cell Reports 2017;8:728–742.ArticlePubMedPMC
- 145. Migdalska-Richards A, Daly L, Bezard E, Schapira AH. Ambroxol effects in glucocerebrosidase and α-synuclein transgenic mice. Ann Neurol 2016;80:766–775.ArticlePubMedPMCPDF
- 146. Migdalska-Richards A, Ko WKD, Li Q, Bezard E, Schapira AHV. Oral ambroxol increases brain glucocerebrosidase activity in a nonhuman primate. Synapse 2017;71:e21967. ArticlePubMedPMCPDF
- 147. Mullin S, Smith L, Lee K, D’Souza G, Woodgate P, Elflein J, et al. Ambroxol for the treatment of patients with Parkinson disease with and without glucocerebrosidase gene mutations: a nonrandomized, noncontrolled trial. JAMA Neurol 2020;77:427–434.ArticlePubMedPMC
- 148. Yap TL, Gruschus JM, Velayati A, Westbroek W, Goldin E, Moaven N, et al. Alpha-synuclein interacts with glucocerebrosidase providing a molecular link between Parkinson and Gaucher diseases. J Biol Chem 2011;286:28080–28088.PubMedPMC
- 149. Yap TL, Jiang Z, Heinrich F, Gruschus JM, Pfefferkorn CM, Barros M, et al. Structural features of membrane-bound glucocerebrosidase and α-synuclein probed by neutron reflectometry and fluorescence spectroscopy. J Biol Chem 2015;290:744–754.ArticlePubMedPMC
- 150. Bae EJ, Yang NY, Song M, Lee CS, Lee JS, Jung BC, et al. Glucocerebrosidase depletion enhances cell-to-cell transmission of α-synuclein. Nat Commun 2014;5:4755.ArticlePubMedPMCPDF
- 151. Gegg ME, Verona G, Schapira AHV. Glucocerebrosidase deficiency promotes release of α-synuclein fibrils from cultured neurons. Hum Mol Genet 2020;29:1716–1728.ArticlePubMedPMCPDF
- 152. Migdalska-Richards A, Wegrzynowicz M, Harrison IF, Verona G, Bellotti V, Spillantini MG, et al. L444P Gba1 mutation increases formation and spread of α-synuclein deposits in mice injected with mouse alphasynuclein pre-formed fibrils. PLoS One 2020;15:e0238075. ArticlePubMedPMC
- 153. Cerri S, Ghezzi C, Ongari G, Croce S, Avenali M, Zangaglia R, et al. GBA mutations influence the release and pathological effects of small extracellular vesicles from fibroblasts of patients with Parkinson’s disease. Int J Mol Sci 2021;22:2215.ArticlePubMedPMC
- 154. Papadopoulos VE, Nikolopoulou G, Antoniadou I, Karachaliou A, Arianoglou G, Emmanouilidou E, et al. Modulation of β-glucocerebrosidase increases α-synuclein secretion and exosome release in mouse models of Parkinson’s disease. Hum Mol Genet 2018;27:1696–1710.ArticlePubMed
- 155. Avenali M, Cerri S, Ongari G, Ghezzi C, Pacchetti C, Tassorelli C, et al. Profiling the biochemical signature of GBA-related Parkinson’s disease in peripheral blood mononuclear cells. Mov Disord 2021;36:1267–1272.ArticlePubMedPMCPDF
- 156. Schapira AH. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol 2008;7:97–109.ArticlePubMed
- 157. Zaltieri M, Longhena F, Pizzi M, Missale C, Spano P, Bellucci A. Mitochondrial dysfunction and α-synuclein synaptic pathology in Parkinson’s disease: who’s on first? Parkinsons Dis 2015;2015:108029.PubMedPMC
- 158. Osellame LD, Rahim AA, Hargreaves IP, Gegg ME, Richard-Londt A, Brandner S, et al. Mitochondria and quality control defects in a mouse model of Gaucher disease--links to Parkinson’s disease. Cell Metab 2013;17:941–953.ArticlePubMedPMC
- 159. Gegg ME, Schapira AH. Mitochondrial dysfunction associated with glucocerebrosidase deficiency. Neurobiol Dis 2016;90:43–50.ArticlePubMedPMC
- 160. Reczek D, Schwake M, Schröder J, Hughes H, Blanz J, Jin X, et al. LIMP2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell 2007;131:770–783.ArticlePubMed
- 161. Michelakakis H, Xiromerisiou G, Dardiotis E, Bozi M, Vassilatis D, Kountra PM, et al. Evidence of an association between the scavenger receptor class B member 2 gene and Parkinson’s disease. Mov Disord 2012;27:400–405.ArticlePubMed
- 162. Hopfner F, Schulte EC, Mollenhauer B, Bereznai B, Knauf F, Lichtner P, et al. The role of SCARB2 as susceptibility factor in Parkinson’s disease. Mov Disord 2013;28:538–540.ArticlePubMed
- 163. Rothaug M, Zunke F, Mazzulli JR, Schweizer M, Altmeppen H, Lullmann-Rauch R, et al. LIMP-2 expression is critical for β- glucocerebrosidase activity and α-synuclein clearance. Proc Natl Acad Sci U S A 2014;111:15573–15578.ArticlePubMedPMC
- 164. Gan-Or Z, Bar-Shira A, Gurevich T, Giladi N, Orr-Urtreger A. Homozygosity for the MTX1 c.184T>A (p.S63T) alteration modifies the age of onset in GBA-associated Parkinson’s disease. Neurogenetics 2011;12:325–332.ArticlePubMedPDF
- 165. Gan-Or Z, Amshalom I, Bar-Shira A, Gana-Weisz M, Mirelman A, Marder K, et al. The Alzheimer disease BIN1 locus as a modifier of GBA-associated Parkinson disease. J Neurol 2015;262:2443–2447.ArticlePubMedPDF
- 166. Schierding W, Farrow S, Fadason T, Graham OEE, Pitcher TL, Qubisi S, et al. Common variants coregulate expression of GBA and modifier genes to delay Parkinson’s disease onset. Mov Disord 2020;35:1346–1356.ArticlePubMedPMCPDF
- 167. Blauwendraat C, Reed X, Krohn L, Heilbron K, Bandres-Ciga S, Tan M, et al. Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 2020;143:234–248.PubMed
- 168. Stoker TB, Camacho M, Winder-Rhodes S, Liu G, Scherzer CR, Foltynie T, et al. A common polymorphism in SNCA is associated with accelerated motor decline in GBA-Parkinson’s disease. J Neurol Neurosurg Psychiatry 2020;91:673–674.ArticlePubMedPMC
- 169. Ysselstein D, Nguyen M, Young TJ, Severino A, Schwake M, Merchant K, et al. LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson’s disease patients. Nat Commun 2019;10:5570.ArticlePubMedPMCPDF
- 170. Krohn L, Özturk TN, Vanderperre B, Ouled Amar Bencheikh B, Ruskey JA, Laurent SB, et al. Genetic, structural, and functional evidence link TMEM175 to synucleinopathies. Ann Neurol 2020;87:139–153.ArticlePubMedPDF
- 171. Jinn S, Blauwendraat C, Toolan D, Gretzula CA, Drolet RE, Smith S, et al. Functionalization of the TMEM175 p.M393T variant as a risk factor for Parkinson disease. Hum Mol Genet 2019;28:3244–3254.ArticlePubMedPMCPDF
- 172. Straniero L, Rimoldi V, Monfrini E, Bonvegna S, Melistaccio G, Lake J, et al. Role of lysosomal gene variants in modulating GBA-associated Parkinson’s disease risk. Mov Disord 2022;37:1202–1210.PubMedPMC
- 173. Weinreb NJ, Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, et al. Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am J Med 2002;113:112–119.ArticlePubMed
- 174. Connock M, Burls A, Frew E, Fry-Smith A, Juarez-Garcia A, McCabe C, et al. The clinical effectiveness and cost-effectiveness of enzyme replacement therapy for Gaucher’s disease: a systematic review. Health Technol Assess 2006;10:iii-iv, ix-136. ArticlePDF
- 175. Brady RO, Yang C, Zhuang Z. An innovative approach to the treatment of Gaucher disease and possibly other metabolic disorders of the brain. J Inherit Metab Dis 2013;36:451–454.ArticlePubMedPMC
- 176. Schueler U, Kaneski C, Murray G, Sandhoff K, Brady RO. Uptake of mannose-terminal glucocerebrosidase in cultured human cholinergic and dopaminergic neuron cell lines. Neurochem Res 2002;27:325–330.PubMed
- 177. Gasca-Salas C, Fernández-Rodríguez B, Pineda-Pardo JA, RodriguezRojas R, Obeso I, Hernández-Fernández F, et al. Blood-brain barrier opening with focused ultrasound in Parkinson’s disease dementia. Nat Commun 2021;12:779.ArticlePubMedPMCPDF
- 178. Meng Y, Reilly RM, Pezo RC, Trudeau M, Sahgal A, Singnurkar A, et al. MR-guided focused ultrasound enhances delivery of trastuzumab to Her2-positive brain metastases. Sci Transl Med 2021;13:eabj4011.ArticlePubMed
- 179. Meng Y, Pople CB, Huang Y, Jones RM, Ottoy J, Goubran M, et al. Putaminal recombinant glucocerebrosidase delivery with magnetic resonance-guided focused ultrasound in Parkinson’s disease: a phase I study. Mov Disord 2022;37:2134–2139.PubMed
- 180. Schiffmann R, Fitzgibbon EJ, Harris C, DeVile C, Davies EH, Abel L, et al. Randomized, controlled trial of miglustat in Gaucher’s disease type 3. Ann Neurol 2008;64:514–522.ArticlePubMedPMC
- 181. Senkevich K, Rudakou U, Gan-Or Z. New therapeutic approaches to Parkinson’s disease targeting GBA, LRRK2 and Parkin. Neuropharmacology 2022;202:108822.ArticlePubMed
- 182. Giraldo P, Andrade-Campos M, Alfonso P, Irun P, Atutxa K, Acedo A, et al. Twelve years of experience with miglustat in the treatment of type 1 Gaucher disease: The Spanish ZAGAL project. Blood Cells Mol Dis 2018;68:173–179.ArticlePubMed
- 183. Sawkar AR, Schmitz M, Zimmer KP, Reczek D, Edmunds T, Balch WE, et al. Chemical chaperones and permissive temperatures alter localization of Gaucher disease associated glucocerebrosidase variants. ACS Chem Biol 2006;1:235–251.ArticlePubMed
- 184. Luan Z, Li L, Higaki K, Nanba E, Suzuki Y, Ohno K. The chaperone activity and toxicity of ambroxol on Gaucher cells and normal mice. Brain Dev 2013;35:317–322.ArticlePubMed
- 185. Sun Y, Liou B, Xu YH, Quinn B, Zhang W, Hamler R, et al. Ex vivo and in vivo effects of isofagomine on acid β-glucosidase variants and substrate levels in Gaucher disease. J Biol Chem 2012;287:4275–4287.ArticlePubMedPMC
- 186. Steet RA, Chung S, Wustman B, Powe A, Do H, Kornfeld SA. The iminosugar isofagomine increases the activity of N370S mutant acid betaglucosidase in Gaucher fibroblasts by several mechanisms. Proc Natl Acad Sci U S A 2006;103:13813–13818.PubMedPMC
- 187. Khanna R, Benjamin ER, Pellegrino L, Schilling A, Rigat BA, Soska R, et al. The pharmacological chaperone isofagomine increases the activity of the Gaucher disease L444P mutant form of beta-glucosidase. FEBS J 2010;277:1618–1638.PubMedPMC
- 188. Tran ML, Génisson Y, Ballereau S, Dehoux C. Second-generation pharmacological chaperones: beyond inhibitors. Molecules 2020;25:3145.ArticlePubMedPMC
- 189. Goldin E, Zheng W, Motabar O, Southall N, Choi JH, Marugan J, et al. High throughput screening for small molecule therapy for Gaucher disease using patient tissue as the source of mutant glucocerebrosidase. PLoS One 2012;7:e29861. ArticlePubMedPMC
- 190. Patnaik S, Zheng W, Choi JH, Motabar O, Southall N, Westbroek W, et al. Discovery, structure-activity relationship, and biological evaluation of noninhibitory small molecule chaperones of glucocerebrosidase. J Med Chem 2012;55:5734–5748.ArticlePubMedPMC
- 191. Aflaki E, Borger DK, Moaven N, Stubblefield BK, Rogers SA, Patnaik S, et al. A new glucocerebrosidase chaperone reduces α-synuclein and glycolipid levels in iPSC-derived dopaminergic neurons from patients with Gaucher disease and parkinsonism. J Neurosci 2016;36:7441–7452.ArticlePubMedPMC
- 192. Aflaki E, Stubblefield BK, Maniwang E, Lopez G, Moaven N, Goldin E, et al. Macrophage models of Gaucher disease for evaluating disease pathogenesis and candidate drugs. Sci Transl Med 2014;6:240ra273.ArticlePubMedPMC
- 193. Mazzulli JR, Zunke F, Tsunemi T, Toker NJ, Jeon S, Burbulla LF, et al. Activation of β-glucocerebrosidase reduces pathological α-synuclein and restores lysosomal function in Parkinson’s patient midbrain neurons. J Neurosci 2016;36:7693–7706.ArticlePubMedPMC
- 194. Marugan JJ, Zheng W, Motabar O, Southall N, Goldin E, Westbroek W, et al. Evaluation of quinazoline analogues as glucocerebrosidase inhibitors with chaperone activity. J Med Chem 2011;54:1033–1058.ArticlePubMedPMC
- 195. Tropak MB, Kornhaber GJ, Rigat BA, Maegawa GH, Buttner JD, Blanchard JE, et al. Identification of pharmacological chaperones for Gaucher disease and characterization of their effects on beta-glucocerebrosidase by hydrogen/deuterium exchange mass spectrometry. Chembiochem 2008;9:2650–2662.ArticlePubMedPMC
- 196. Zheng J, Chen L, Schwake M, Silverman RB, Krainc D. Design and synthesis of potent quinazolines as selective β-glucocerebrosidase modulators. J Med Chem 2016;59:8508–8520.ArticlePubMedPMC
- 197. Zheng J, Jeon S, Jiang W, Burbulla LF, Ysselstein D, Oevel K, et al. Conversion of quinazoline modulators from inhibitors to activators of β- glucocerebrosidase. J Med Chem 2019;62:1218–1230.ArticlePubMedPMC
- 198. Burbulla LF, Jeon S, Zheng J, Song P, Silverman RB, Krainc D. A modulator of wild-type glucocerebrosidase improves pathogenic phenotypes in dopaminergic neuronal models of Parkinson’s disease. Sci Transl Med 2019;11:eaau6870.ArticlePubMedPMC
- 199. Maegawa GH, Tropak MB, Buttner JD, Rigat BA, Fuller M, Pandit D, et al. Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J Biol Chem 2009;284:23502–23516.ArticlePubMedPMC
- 200. Narita A, Shirai K, Itamura S, Matsuda A, Ishihara A, Matsushita K, et al. Ambroxol chaperone therapy for neuronopathic Gaucher disease: a pilot study. Ann Clin Transl Neurol 2016;3:200–215.ArticlePubMedPMCPDF
- 201. Artusi CA, Dwivedi AK, Romagnolo A, Pal G, Kauffman M, Mata I, et al. Association of subthalamic deep brain stimulation with motor, functional, and pharmacologic outcomes in patients with monogenic Parkinson disease: a systematic review and meta-analysis. JAMA Netw Open 2019;2:e187800. ArticlePubMedPMC
- 202. Lythe V, Athauda D, Foley J, Mencacci NE, Jahanshahi M, Cipolotti L, et al. GBA-associated Parkinson’s disease: progression in a deep brain stimulation cohort. J Parkinsons Dis 2017;7:635–644.ArticlePubMed
- 203. Mangone G, Bekadar S, Cormier-Dequaire F, Tahiri K, Welaratne A, Czernecki V, et al. Early cognitive decline after bilateral subthalamic deep brain stimulation in Parkinson’s disease patients with GBA mutations. Parkinsonism Relat Disord 2020;76:56–62.ArticlePubMed
- 204. Pal G, Mangone G, Hill EJ, Ouyang B, Liu Y, Lythe V, et al. Parkinson disease and subthalamic nucleus deep brain stimulation: cognitive effects in GBA mutation carriers. Ann Neurol 2022;91:424–435.PubMedPMC
REFERENCES
Figure & Data
References
Citations
- A Comparative Biochemical and Pathological Evaluation of Brain Samples from Knock-In Murine Models of Gaucher Disease
Makaila L. Furderer, Bahafta Berhe, Tiffany C. Chen, Stephen Wincovitch, Xuntian Jiang, Nahid Tayebi, Ellen Sidransky, Tae-Un Han
International Journal of Molecular Sciences.2024; 25(3): 1827. CrossRef - Towards a Global View of Parkinson's Disease Genetics
Marzieh Khani, Catalina Cerquera‐Cleves, Mariam Kekenadze, Peter Wild Crea, Andrew B. Singleton, Sara Bandres‐Ciga
Annals of Neurology.2024;[Epub] CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite