Paroxysmal dyskinesias are a rare group of movement disorders that are defined by involuntary intermittent movements that may include any combination of dystonia, chorea, or athetosis. In 1885, Gower published a book that included the description of two pediatric cases with movement-induced seizures. Overtime, reports of episodes being preceded by an aura, triggered by fatigue or exercise, and lasting only minutes were published [1-3].
Paroxysmal kinesigenic dyskinesia (PKD) may be primarily caused by an abnormality of the PRRT2 gene, which is inherited in an autosomal dominant fashion [4]. Secondary basis, such as occurring due to a lesion, has been documented in strokes and multiple sclerosis, which is often manifested as unilateral movements that are contralateral to the side of the lesion. This case report is unique, since concussions have not been previously reported to induce PKD.
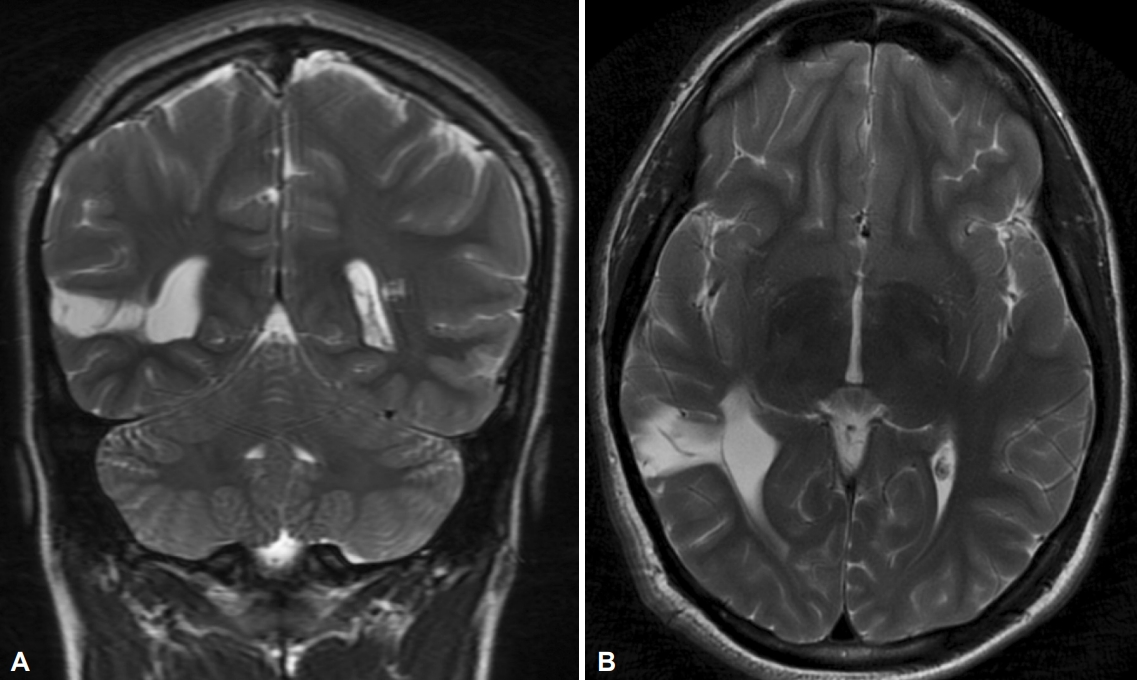
The patient is a 16-year-old male who suffered a concussion while playing soccer. He did not have loss of consciousness with the injury and had headaches several months following the concussion. The headaches eventually subsided. There was significant past medical history involving a right craniotomy as an infant for resection of a choroid plexoma in the right hemisphere. The choroid plexoma was found during a prenatal ultrasound, since he had no symptoms of increased intracranial pressure or abnormalities during examination. The patient had no history of seizures. MRI of the brain following the concussion demonstrated right temporal encephalomalacia, consistent with a prior craniotomy. No other lesion was present (Figure 1). Physical examination did not reveal any left sided weakness. However, he was right handed with better dexterity in the right hand. The patient returned several months after his head injury and reported new onset episodes of involuntary bilateral upper extremity dystonic movements, in which the left arm had larger and more noticeable movements than the right arm. He denied an aura or other abnormal sensations. These episodes occurred with sudden movement and stimulation, such as when his soccer coach called him from the bench to play in the game and he suddenly stood up and ran. Movements lasted a few seconds, and he would often use his right arm to hold the left arm to make movements less noticeable. If he was startled at home or at school, asymmetric dystonic movements of his arms also occurred. These events occurred multiple times per day, or as often as he was startled.
Neurological examination revealed symmetric reflexes and strength and no pronator drift. Patient did not toe walk or drag the left leg. Tone was also symmetric, but slight decreased range of motion was noted in the left ankle. No clonus was present.
The family reported that his father’s uncle had similar episodes as a teenager. The paternal great uncle was treated with phenobarbital but self-discontinued the medication, since it did not improve the movements and caused drowsiness. However, his movements resolved without intervention by the age of 30. The great uncle did not have genetic testing performed. There was no family history of focal dystonia or seizures. The mother reported occasional migraine headaches.
For our case patient, oxcarbazepine was initiated at a moderate dosage of 300 mg twice a day. Our patient’s movements resolved as long as he was compliant with the medication. He was able to continue playing soccer and reported no dystonic movements, even with startling, while taking the medication. Routine interictal EEG was normal. The family declined genetic testing.
Primary kinesigenic dyskinesia is defined as sudden episodes that are often preceded by an aura and include dystonia, chorea or athetosis, and can be bilateral, unilateral or asymmetric. We feel that our patient had primary PKD, since the movements were bilateral and there was also a family history of PKD. Presence of more pronounced movements in the left arm was likely due to the contralateral right temporal lobe encephalomalacia. The patient’s recent concussion seemed to lower the patient’s threshold for primary PKD. If the PKD was secondary from encephalomalacia, the PKD likely would have occurred sooner following the craniotomy and would be unilateral. Most often, secondary PKD is unilateral, and in our case, his movements were mainly bilateral. Genetic testing was not performed in our patient, which is unfortunate, since this could have identified the gene. The PRRT2 gene has been definitively linked as a singular etiology of PKD, and the PRRT2 gene is highly expressed in the basal ganglia. The PRRT2 gene is understood to be a part of the calcium mediated SNAP25 neuronal exocytosis at the presynaptic membrane [5].
Unfortunately, we were not able to obtain genetic testing in our patient. If testing had been positive for the PRRT2 gene, then we could clearly assume his movements were genetic in etiology. Likewise, a negative genetic finding would have suggested the acute brain injury to be the cause. However, based on family history of similar movements (and treatment response), age at presentation, stability of radioimaging and presence of bilateral movements (asymmetry likely due to underlying weakness of arm), we feel that this patient does have primary (genetic) PKD.
We describe a rare case of PKD with initiation induced by concussion. Concussion or post-concussive syndrome has not been reported to initiate this disorder. However, with the family history of PKD, we feel that the patient’s concussion predisposed him to manifest his genetic tendency for PKD was not the sole etiology for PKD. He improved with oxcarbazepine, which is similar to prior reports showing improvement with carbazepine [6]. Another interesting aspect is his pre-concussive right encephalomalacia which caused dyskinesias to be more severe in the left arm.
Notes
-
Conflicts of Interest
The authors have no financial conflicts of interest.
Figure 1.Right temporal encephalomalacia corresponding to area of removal of prior choroid plexus tumor. A: Coronal view. B: Axial view.
REFERENCES
- 1. Johnston M, Adams Jr H, and Fatemi A. Neurobiology of Disease. 2nd ed. New York: Oxford University Press; 2016.
- 2. Lehman R, Mink J. Paroxysmal dyskinesias. J Pediatr Neurol 2010;8:65–67.Article
- 3. Gardiner AR, Jaffer F, Dale RC, Labrum R, Erro R, Meyer E, et al. The clinical and genetic heterogeneity of paroxysmal dyskinesias. Brain 2015;138(Pt 12):3567–3580.ArticlePubMedPMCPDF
- 4. Wang JL, Cao L, Li XH, Hu ZM, Li JD, Zhang JG, et al. Identification of PRRT2 as the causative gene of paroxysmal kinesigenic dyskinesias. Brain 2011;134(Pt 12):3493–3501.ArticlePubMedPDF
- 5. Unterberger I, Trinka E. Diagnosis and treatment of paroxysmal dyskinesias revisited. Ther Adv Neurol Disord 2008;1:4–11.ArticlePubMedPMC
- 6. Yang Y, Su Y, Guo Y, Ding Y, Xu S, Jiang Y, et al. Oxcarbazepine versus carbamazepine in the treatment of paroxysmal kinesigenic dyskinesia. Int J Neurosci 2012;122:719–722.ArticlePubMed
Citations
Citations to this article as recorded by

- Concussion-Induced Oromandibular Dyskinesia
Brenda Zhang, Joseph P Settineri, Alan Chajet, Purushothaman Muthukanagaraj
Cureus.2023;[Epub] CrossRef - Recommendations for the diagnosis and treatment of paroxysmal kinesigenic dyskinesia: an expert consensus in China
Li Cao, Xiaojun Huang, Ning Wang, Zhiying Wu, Cheng Zhang, Weihong Gu, Shuyan Cong, Jianhua Ma, Ling Wei, Yanchun Deng, Qi Fang, Qi Niu, Jin Wang, Zhaoxia Wang, You Yin, Jinyong Tian, Shufen Tian, Hongyan Bi, Hong Jiang, Xiaorong Liu, Yang Lü, Meizhen Sun
Translational Neurodegeneration.2021;[Epub] CrossRef
 E-submission
E-submission


 PubReader
PubReader ePub Link
ePub Link Cite
Cite
