E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 11(2); 2018 > Article
-
Case Report
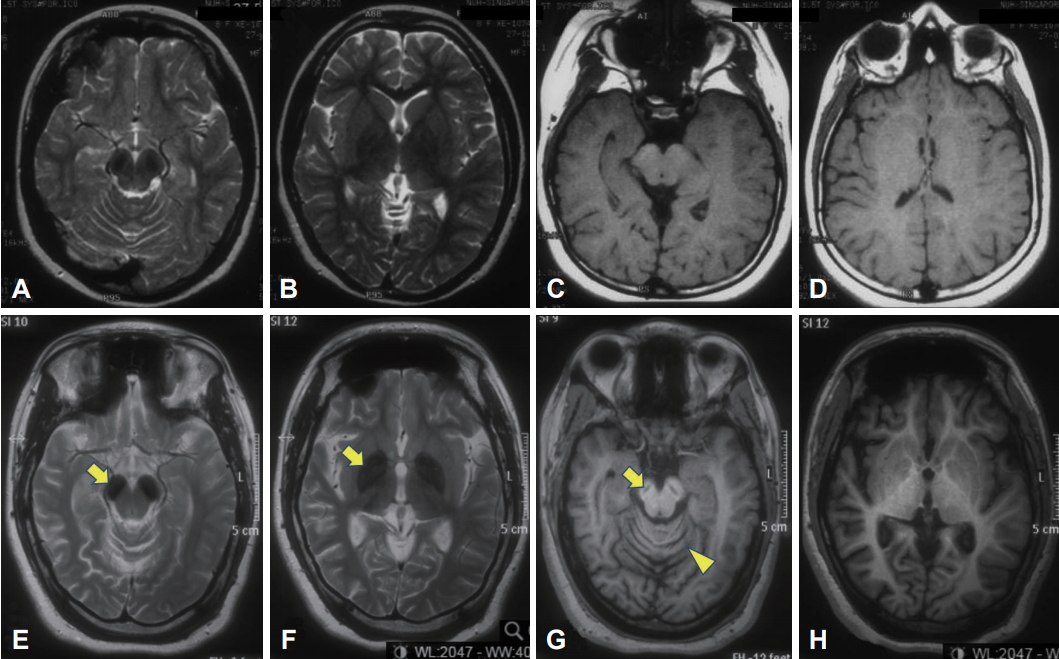
A Patient with Beta-Propeller Protein-Associated Neurodegeneration: Treatment with Iron Chelation Therapy -
Shen-Yang Lim1,2
 , Ai Huey Tan1,2, Azlina Ahmad-Annuar3, Susanne A. Schneider4, Ping Chong Bee5, Jia Lun Lim2,3, Norlisah Ramli6, Mohamad Imran Idris1
, Ai Huey Tan1,2, Azlina Ahmad-Annuar3, Susanne A. Schneider4, Ping Chong Bee5, Jia Lun Lim2,3, Norlisah Ramli6, Mohamad Imran Idris1 -
Journal of Movement Disorders 2018;11(2):89-92.
DOI: https://doi.org/10.14802/jmd.17082
Published online: May 30, 2018
1Divisions of Neurology, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia
2The Mah Pooi Soo & Tan Chin Nam Centre for Parkinson’s & Related Disorders, University of Malaya, Kuala Lumpur, Malaysia
3Department of Biomedical Science, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia
4Department of Neurology, University of Munich, Munich, Germany
5Divisions of Haematology, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia
6Divisions of Neuroradiology, Faculty of Medicine, University of Malaya, Kuala Lumpur, Malaysia
- Corresponding author: Shen-Yang Lim, MBBS, MD, FRACP, FASc, https://orcid.org/0000-0002-6942-2522 Neurology Laboratory, Level 6 (South Block), University of Malaya Medical Centre, Kuala Lumpur 50603, Malaysia / Tel: +60-3-7949-2891 / Fax: +60- 3-7949-4613 / E-mail: limshenyang@gmail.com
Copyright © 2018 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
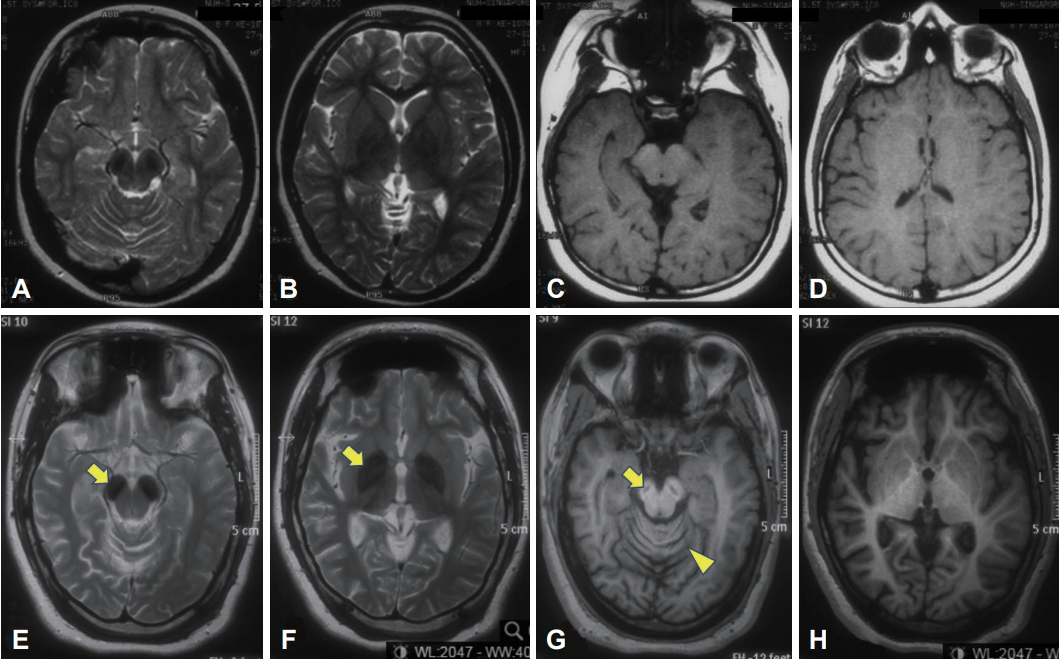
Figure & Data
References
Citations
- Lipid droplet accumulation in Wdr45-deficient cells caused by impairment of chaperone-mediated autophagic degradation of Fasn
Qiuhong Xiong, Huimin Sun, Yanlin Wang, Qian Xu, Yu Zhang, Mei Xu, Zhonghua Zhao, Ping Li, Changxin Wu
Lipids in Health and Disease.2024;[Epub] CrossRef - Quantitative retrospective natural history modeling of WDR45-related developmental and epileptic encephalopathy – a systematic cross-sectional analysis of 160 published cases
Afshin Saffari, Julian Schröter, Sven F. Garbade, Julian E. Alecu, Darius Ebrahimi-Fakhari, Georg F. Hoffmann, Stefan Kölker, Markus Ries, Steffen Syrbe
Autophagy.2022; 18(7): 1715. CrossRef - Cerebral Iron Deposition in Neurodegeneration
Petr Dusek, Tim Hofer, Jan Alexander, Per M. Roos, Jan O. Aaseth
Biomolecules.2022; 12(5): 714. CrossRef - Interactions of dopamine, iron, and alpha-synuclein linked to dopaminergic neuron vulnerability in Parkinson's disease and Neurodegeneration with Brain Iron Accumulation disorders
Rachel M. Wise, Annika Wagener, Urban M. Fietzek, Thomas Klopstock, Eugene V. Mosharov, Fabio A. Zucca, David Sulzer, Luigi Zecca, Lena F. Burbulla
Neurobiology of Disease.2022; 175: 105920. CrossRef -
WDR45 variants cause ferrous iron loss due to impaired ferritinophagy associated with nuclear receptor coactivator 4 and WD repeat domain phosphoinositide interacting protein 4 reduction
Kiwako Tsukida, Shin-ichi Muramatsu, Hitoshi Osaka, Takanori Yamagata, Kazuhiro Muramatsu
Brain Communications.2022;[Epub] CrossRef - Iron Chelation in Movement Disorders: Logical or Ironical
Dinkar Kulshreshtha, Jacky Ganguly, Mandar Jog
Canadian Journal of Neurological Sciences / Journal Canadien des Sciences Neurologiques.2021; : 1. CrossRef - Emerging Disease-Modifying Therapies in Neurodegeneration With Brain Iron Accumulation (NBIA) Disorders
Vassilena Iankova, Ivan Karin, Thomas Klopstock, Susanne A. Schneider
Frontiers in Neurology.2021;[Epub] CrossRef - Consensus clinical management guideline for beta‐propeller protein‐associated neurodegeneration
Jenny L Wilson, Allison Gregory, Manju A Kurian, Ittai Bushlin, Fanny Mochel, Lisa Emrick, Laura Adang, Penelope Hogarth, Susan J Hayflick
Developmental Medicine & Child Neurology.2021; 63(12): 1402. CrossRef - WDR45, one gene associated with multiple neurodevelopmental disorders
Yingying Cong, Vincent So, Marina A. J. Tijssen, Dineke S. Verbeek, Fulvio Reggiori, Mario Mauthe
Autophagy.2021; 17(12): 3908. CrossRef - Towards Precision Therapies for Inherited Disorders of Neurodegeneration with Brain Iron Accumulation
Robert V.V. Spaull, Audrey K.S. Soo, Penelope Hogarth, Susan J. Hayflick, Manju A. Kurian
Tremor and Other Hyperkinetic Movements.2021;[Epub] CrossRef - The roles of iron and HFE genotype in neurological diseases
Yunsung Kim, James R. Connor
Molecular Aspects of Medicine.2020; 75: 100867. CrossRef - The Contribution of Iron to Protein Aggregation Disorders in the Central Nervous System
Karina Joppe, Anna-Elisa Roser, Fabian Maass, Paul Lingor
Frontiers in Neuroscience.2019;[Epub] CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite