E-submission
E-submission
Articles
- Page Path
- HOME > J Mov Disord > Volume 2(1); 2009 > Article
-
Original Article
Comparison of Cerebral Glucose Metabolism between Possible and Probable Multiple System Atrophy - Kyum-Yil Kwona, Jae Seung Kimb, Ki Chun Imb, Myoung Chong Leea, Sun Ju Chunga
-
Journal of Movement Disorders 2009;2(1):22-28.
DOI: https://doi.org/10.14802/jmd.09006
Published online: April 30, 2009
aCenter for Parkinsonism and Other Movement Disorders, Departments of Neurology, University of Ulsan College of Medicine, Seoul, Korea
bNuclear Medicine, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
- Corresponding author: Sun Ju Chung, MD, PhD, Center for Parkinsonism and Other Movement Disorders, Department of Neurology, Asan Medical Center, University of Ulsan College of Medicine, 388-1 Poongnap-dong, Songpa-gu, Seoul 138-736, Korea, Tel +82-2-3010-3988, Fax +82-2-474-4691, E-mail sjchung@amc.seoul.kr
• Received: February 14, 2009 • Revised: March 20, 2009 • Accepted: March 14, 2009
Copyright © 2009 The Korean Movement Disorder Society
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 10,143 Views
- 85 Download
- 2 Crossref
ABSTRACT
-
Background:
- To investigate the relationship between presenting clinical manifestations and imaging features of multisystem neuronal dysfunction in MSA patients, using 18F-fluorodeoxyglucose positron emission tomography (18F-FDG PET).
-
Methods:
- We studied 50 consecutive MSA patients with characteristic brain MRI findings of MSA, including 34 patients with early MSA-parkinsonian (MSA-P) and 16 with early MSA-cerebellar (MSA-C). The cerebral glucose metabolism of all MSA patients was evaluated in comparison with 25 age-matched controls. 18F-FDG PET results were assessed by the Statistic Parametric Mapping (SPM) analysis and the regions of interest (ROI) method.
-
Results:
- The mean time from disease onset to 18F-FDG PET was 25.9±13.0 months in 34 MSA-P patients and 20.1±11.1 months in 16 MSA-C patients. Glucose metabolism of the putamen showed a greater decrease in possible MSA-P than in probable MSA-P (p=0.031). Although the Unified Multiple System Atrophy Rating Scale (UMSARS) score did not differ between possible MSA-P and probable MSA-P, the subscores of rigidity (p=0.04) and bradykinesia (p= 0.008) were significantly higher in possible MSA-P than in probable MSA-P. Possible MSA-C showed a greater decrease in glucose metabolism of the cerebellum than probable MSA-C (p=0.016).
-
Conclusions:
- Our results may suggest that the early neuropathological pattern of possible MSA with a predilection for the striatonigral or olivopontocerebellar system differs from that of probable MSA, which has prominent involvement of the autonomic nervous system in addition to the striatonigral or olivopontocerebellar system.
- Multiple system atrophy (MSA) is a sporadic, relentlessly progressive neurodegenerative disorder characterized clinically by various combinations of parkinsonism, cerebellar dysfunction, autonomic dysfunction, and corticospinal tract dysfunction.1 Despite considerable recent advances in research into neurodegenerative disorders, the diagnosis of MSA still rests on the clinical history and neurological examination, and several investigations have been proposed to aid in the differential diagnosis of this disease from Parkinson’s disease or other atypical parkinsonisms.2–8
- Two types of MSA have been distinguished in terms of clinicopathological correlations.1,9 Parkinsonian type MSA (MSA-P), predominantly displaying parkinsonian features, is correlated with pathological involvement of the striatonigral system, whereas cerebellar type MSA (MSA-C), predominantly revealing cerebellar ataxia, is associated with that of the olivopontocerebellar system. MSA-P patients present initially with extrapyramidal features, and commonly progress to develop autonomic disturbances, cerebellar ataxia, or both. Conversely, MSA-C patients who begin with cerebellar dysfunction often progress to develop parkinsonian or autonomic dysfunction, or both.
- The Consensus criteria are currently widely used for a clinical diagnosis of MSA.1 These criteria specify three diagnostic categories of increasing certainty: possible, probable, and definite MSA. The diagnosis of possible and probable MSA is based on the presence of clinical features listed in the Consensus criteria. A retrospective evaluation of the Consensus criteria on pathologically proven cases showed excellent positive predictive values for both possible (93%) and probable MSA (100%) at the first clinic visit,10 although the sensitivity for probable MSA was poor.
- The rate of progression in MSA may vary considerably between individual MSA patients9,11 and predictors of progression of the disease remain unclear. Although neuropathological characteristics of the striatonigral system and olivopontocerebellar system in MSA have been described,12,13 their correlation with pathological changes of the autonomic nervous system remains to be clarified. We therefore investigated the cerebral glucose metabolism of patients with possible MSA who had no or mild autonomic dysfunction and those with probable MSA who had severe autonomic dysfunction.
Introduction
- Patients
- We studied 34 patients with MSA-P (possible 10, probable 24) and 16 with MSA-C (possible 10, probable 6) who underwent 18F-fluorodeoxyglucose (FDG) PET studies within 5 years after disease onset at Asan Medical Center, South Korea, between January 2004 and July 2007. Clinical diagnoses of MSA were assessed by experienced movement disorder experts (M.C.L and S.J.C). All MSA patients were diagnosed on the basis of Consensus statement for the diagnosis of MSA1 and were classified as having MSA-P or MSA-C. For the patients with possible MSA as well as those with probable MSA, we only included patients who had characteristic brain MRI findings of MSA to increase the probability of clinical diagnosis of MSA.8 A detailed clinical history was obtained from each patient, and careful neurological examination was performed to exclude the presence of other atypical parkinsonism such as progressive supranuclear gaze palsy, corticobasal degeneration, or frontotemporal dementia. Patients excluded from this study included those with symptoms suggestive of Parkinson’s disease, other atypical parkinsonism, secondary parkinsonism, dementia, other significant neurologic diseases, psychiastric disorders, or severe systemic illness. In all patients, PET studies were performed after written informed consents.
- Clinical assessment
- Patients were assessed on disease duration, Unified Parkinson’s Disease Rating Scale (UPDRS),14 Unified Multiple System Atrophy Rating Scale (UMSARS),15 and Hoehn and Yahr stage. The tremor score was defined as the sum of the motor subscore of UPDRS part III items 20 (resting type) and 21 (postural type) in all four limbs and face (score range, 0–28). The rigidity score was defined as the sum of the motor subscore of UPDRS part III item 22 in all four limbs and neck (score range, 0–20). The bradykinesia score was defined as the sum of the motor subscore of UPDRS part III items 23–26 and 31 in all four limbs and body (score range, 0–36). The ‘axial’ subscore was defined as the sum of the motor subscores for speech, facial expression, chin-tongue tremor, neck rigidity, arising from a chair, posture, and body bradykinesia (items 18–20, 22, 27, 28, and 31 of UPDRS part III)(score range, 0–28). The postural instability and gait difficulty (PIGD) subscore was defined as the sum of the motor subscores of the UPDRS part III items 13, 15, 29, and 30 (score range, 0–16).
- Patients with MSA-P or MSA-C were divided into two groups according to disease duration (≤2 vs. >2 years), Hoehn and Yahr stage (≤2 vs. >2), and diagnostic category (possible vs. probable MSA).
- 18F-FDG PET scans and statistical parametric mapping analysis
- 18F-FDG PET was performed in all 50 patients with MSA, all of whom had fasted for at least 6 hours before the scan. Transmission scan of 5 min using a 68Ge rotating pin source and emission scan of 15 min were acquired using ECAT HR+scanner (Siemens Medical Systems, Hoffman estate, IL, USA) 40 min after intravenous injection of 370 MBq 18F-FDG. The system provides 63 planes (2.46 mm slice thickness) and a 15.5 cm longitudinal field view. After a 5-min transmission scan for segmented attenuation correction with a rotating 68Ge source, an emission scan was acquired for 15 min in three-dimensional mode. All emission images were reconstructed with ordered subset expectation maxi-mization (OSEM) using 16 subsets and six iterations. 18F-FDG PET images were normalized using SPM2 (Wellcome Functional Imaging Laboratory) in conjunction with MATLAB version 6.5 (The Mathworks Inc., USA) running on Windows NT. After converting the data from DICOM file into Analyze format, PET images obtained from patients with MSA symptoms that were worse on the right side of the body were flipped to the contralateral side (right) for the group analysis of ipsilateral and contralateral brain glucose metabolism. The images from each subject were spatially transformed to a generic SPM2 PET template.
- Twenty-five normal volunteers (mean age, 60.4±8.4 years; 14 males and 11 females) were used as controls in the SPM analysis. All normal volunteers had no history of neurological or psychological disease, and they did not take any medication. Grouped 18F-FDG PET results were analyzed for significant glucose hypermetabolism or hypometabolism of the putamen, pons, and cerebellum using SPM at a false discovery rate (FDR) corrected statistical threshold of p<0.01 (minimum cluster size ≥50).
- Regions of interest analysis of 18F-FDG PET
- To confirm the SPM results, binary comparisons according to disease duration, Hoehn and Yahr stage, and diagnostic categories of MSA (possible vs. probable) were also investigated using the ROI method. We used the macroscopic anatomical parcellation method with an Automated Anatomical Labeling template16 and ROI was automatically defined among the 90 three-dimensional anatomical volumes of interest on the basis of SPM results. The average glucose metabolic activity of each ROI (putamen and cerebellum) was calculated by total glucose activity of the ROI divided by its volume (MBq/cc), using the in-house program developed with Interactive Data Language (IDL, version 5.5). For quantitative analysis, the individual value of the ROI was normalized to the global brain activity to correct for intraparticipant or interparticipant variability in glucose metabolism and 18F-FDG dose.
- Statistical analysis
- Contingency tables were analyzed using the chi-square test. The Mann-Whitney U test was used to compare non-parametric continuous variables for different groups. For the analysis of SPM data, a two-sided t-test was performed to assess the group comparison. The analysis produced a t statistic for each voxel and t values were transformed to the standard distribution (Z scores). SPSS for Windows (version 12.0, SPSS Inc.) was used for all statistical analyses and p values <0.05 were regarded as statistically significant.
Patients and Methods
- Clinical characteristics of patients
- The clinical characteristics of the patients with MSA-P and MSA-C are summarized in Table 1. Patient age, disease duration, and time interval from the onset of clinical motor symptoms to the 18F-FDG PET study did not differ significantly between possible and probable MSA subtypes.
- 18F-FDG PET analysis of MSA-P using SPM and ROI methods
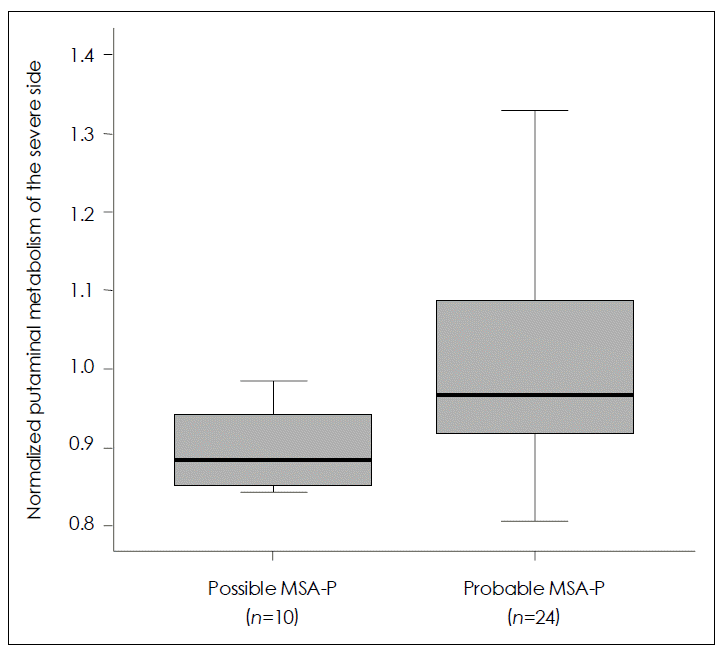
- SPM analysis of the MSA-P group in comparison with normal controls (Figure 1) revealed a greater decrease in glucose metabolism visually in the putamen contralateral to the body side with worse parkinsonism in patients with possible MSAP than in those with probable MSA-P. ROI method analysis (Figure 2) also showed a greater decrease in glucose metabolism in the putamen contralateral to the body side with worse parkinsonism in patients with possible MSA-P (median, 0.88; range, 0.84–0.98) than in those with probable MSA-P (median, 0.97; range, 0.81–1.32)(p=0.018).
- There was no significant difference in glucose metabolism between MSA-P patients with disease duration of ≤2 years (median, 0.95; range, 0.81–1.33) and those with disease duration of >2 years (median, 0.93; range, 0.84–1.18)(p=0.584). In addition, in patients with MSA-P, glucose metabolism did not differ between patients with Hoehn and Yahr stage ≤2 (median, 0.93; range, 0.81–1.14) and those with Hoehn and Yahr stage >2 (median, 0.94; range, 0.84–1.33)(p=0.796).
- 18F-FDG PET analysis of MSA-C using SPM and ROIs methods
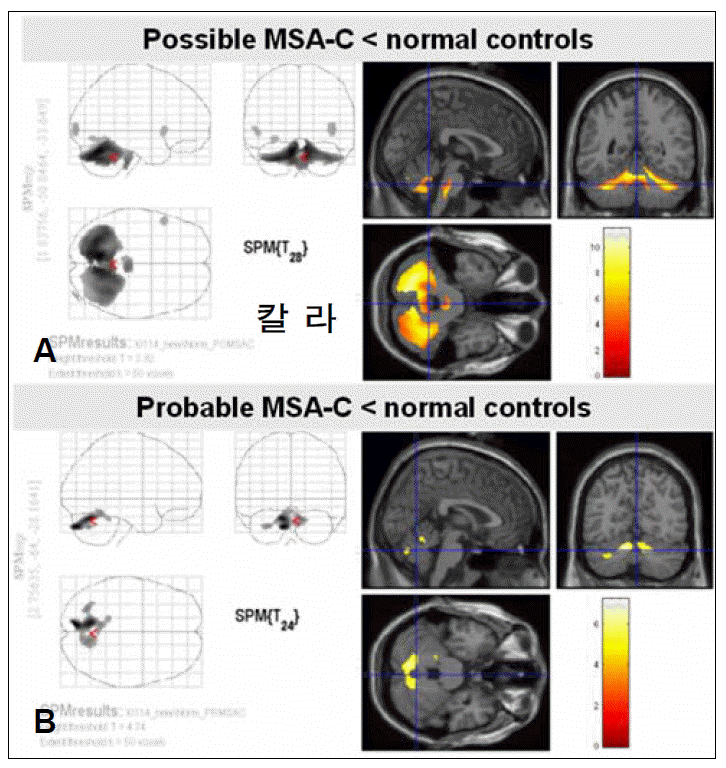
- SPM analysis of the MSA-C group in comparison with normal controls (Figure 3) revealed a greater decrease in glucose metabolism visually in the cerebellum in patients with possible MSA-C than in those with probable MSA-C. ROI method analysis (Figure 4) showed a greater decrease in glucose metabolism in the cerebellum in patients with possible MSA-C (median, 0.61; range, 0.53–0.69) than in those with probable MSA-C (median, 0.67; range, 0.62–0.69)(p= 0.016).
- In patients with MSA-C, glucose metabolism did not differ between patients with disease duration of ≤2 years (median, 0.62; range, 0.53–0.69) and those with disease duration >2 years (median, 0.63; range, 0.58–0.69)(p=0.798), nor did glucose metabolism differ between patients with Hoehn and Yahr stage ≤2 (median, 0.66; range, 0.61–0.69) and those with Hoehn and Yahr stage >2 (median, 0.62; range, 0.53–0.69)(p= 0.263).
- Analysis of clinical scales in patients with MSA-P
- UPDRS and UMSARS were evaluated in 15 patients with MSA-P (Table 2). There was no difference between patients with possible and probable MSA-P in motor UPDRS III subscores and total UPDRS scores. UMSARS Part II score did not differ between patients with probable and possible MSA-P. However, patients with possible MSA-P showed higher subscores for rigidity and bradykinesia of the body side with worse parkinsonism than those with probable MSA-P. There was no difference between patients with possible and probable MSA-P for tremor subscore, axial subscore, PIGD subscore, Hoehn and Yahr stage, and UMSARS Part IV scores.
Results
- In the present study, patients with possible MSA who had no or mild autonomic dysfunction showed a greater decrease in cerebral glucose metabolism than those with probable MSA who had severe autonomic dysfunction. In addition, the scores for rigidity and bradykinesia of the body side with worse parkinsonism in patients with possible MSA-P were higher than in those with probable MSA-P. These imaging and clinical findings suggest that the pattern of pathological involvement of early probable MSA, predominantly involving autonomic systems, differs from that of early possible MSA, mainly involving the striatonigral or olivopontocerebellar system.
- Our results are in line with previous suggestions that motor progression in MSA is unrelated to the presence of additional cerebellar symptoms and pyramidal signs at baseline,17 and that progression of MSA is not uniform but depends on the presence or absence of distinct core features.18 In the present study, although patients with possible MSA had no or mild autonomic dysfunction, they showed a greater decrease in glucose metabolism in the putamen or cerebellum than those with probable MSA, suggesting the independent pathological involvement and progression of the autonomic nervous system compared with the striatonigral or olivopontocerebellar system.
- To date, the patterns and progression of pathological changes of the brain and autonomic nervous system in MSA remain to be clarified. The variable combinations of clinical manifestations of MSA suggest heterogeneous involvement of the nervous system, including the striatonigral system, olivopontocerebellar system, corticospinal tract, and autonomic nervous system.11–13 Clinically, a diagnosis of MSA is made based on the Consensus criteria1 and MSA categories, including MSA-P and MSA-C, are assessed according to the predominant motor presentation. In reviewing the medical records of our patients, most patients with probable MSA developed autonomic failure earlier than parkinsonism or cerebellar dysfunction, whereas all patients with possible MSA presented with motor symptoms earlier than autonomic failure. Therefore, the diagnostic categories of MSA suggested by the Consensus criteria1 may not be sufficient to recognize the characteristics of heterogeneous clinical presentations and the progression of MSA.
- The Consensus criteria use the terms “criteria” and “features” in a specific and precise way.1 Compared with Quinn possible and probable categories, the Consensus probable category showed considerably lower sensitivities than the possible category.10 The Consensus possible category of MSA requires a greater number of dysfunction items as options, whereas the probable category requires fewer, but more obligatory, items. The “criteria” items in Consensus criteria of MSA are stricter than the “feature” items of four domains that include autonomic and urinary dysfunction, parkinsonism, cerebellar dysfunction, and corticospinal tract dysfunction. The Consensus criteria of MSA,1 as well as the modified Quinn criteria,9 were developed on the basis of expert opinion rather than a robust empirical evidence-based or clinicopathological correlation. Because there is no one set of ideal criteria of MSA, the Consensus possible and probable criteria can be used depending on the clinical and research objectives. The Consensus possible criteria can be used in a clinical trial that requires high sensitivity at the expense of positive predictive value, such as a pragmatic trial of a cheap, safe and potentially effective therapy that requires initiation early in the natural course of the disease. In the present study, 20 patients with possible MSA who had characteristic brain MRI findings of MSA were evaluated.
- Although several highly regarded studies have described the clinico-pathological correlations in patients with MSA,12,13,19,20 most of those studies have been retrospective and used tissue from brain banks, which might cause selection bias. These previous postmortem studies had limitations in demonstrating the relation between key pathological changes and the rate of MSA progression. 18F-FDG PET provides a sensitive means of detecting and characterizing the regional changes in brain metabolism associated with MSA,7,8,21–24 and can be of diagnostic value in patients with early-stage MSA and in objectively following disease progression. Using 18F-FDG PET, we aimed to investigate the relationship between regional brain metabolism and various combinations of clinical manifestations in patients with early-stage MSA. In the present study, MSA patients had mean disease durations of less than 3 years; the patients with probable MSA had obvious autonomic dysfunction and showed a lesser degree of cerebral glucose hypometabolism than those with possible MSA who had no or mild autonomic dysfunction. Our results suggest that the severity of pathological involvement of the autonomic nervous system in MSA may not be correlated with the severity of striatonigral or olivopontocerebellar degeneration.
- Patients with MSA-P may have atypical features compared with those with Parkinson’s disease, such as poor response to levodopa,19,25 rapid disease progression,17,20,26–28 jerky postural tremor,9 or focal reflex myoclonus.29 However, poor diagnostic sensitivity and accuracy of MSA among movement disorder specialists have been reported.10,30,31 In the present study, patients with possible MSA showed a greater decrease in glucose metabolism in the putamen or cerebellum than those with probable MSA, which has clinical implications. Indeed, the identification of patients with only “possible” items of MSA is challenging, in view of the lack of definite diagnostic tests. Moreover, neuropathological studies have shown that even signs and symptoms that are considered diagnostic of Parkinson’s disease may be found in patients ultimately recognized to have MSA.25,26,32 Therefore, the 18F-FDG PET findings in our patients with possible MSA suggest that 18F-FDG PET may be a highly useful imaging tool in confirming the clinical suspicion of early-stage MSA.
- The present study had limitations. First, 18F-FDG PET was evaluated in patients clinically diagnosed with MSA, but in the absence of pathological information. Second, semi-quantitative comparison between motor features and 18F-FDG PET findings was not performed.
- In conclusion, patients with early-staged possible MSA showed a greater decrease in cerebral glucose metabolism of the putamen or cerebellum than those with early-staged probable MSA. Our results suggest that the early neuropathological pattern of possible MSA with a predilection for the striatonigral or olivopontocerebellar system may differ from that of probable MSA with predominant involvement of the autonomic nervous system. A larger, well-designed prospective study with neuropathological data is required to confirm our findings.
Discussion
- This study was supported by a grant (2007-416) from the Asan Institute for Life Sciences, Seoul, Korea.
Acknowledgments
Figure 1.Statistical parametric mapping (SPM) analysis of possible and probable Parkinsonian type multiple system atrophy (MSA-P) by 18F-FDG PET. A greater decrease in glucose metabolism is revealed in the putamen in patients with possible MSA-P (A) than in those with probable MSA-P (B), at a false discovery rate (FDR) corrected statistical threshold of p<0.01.
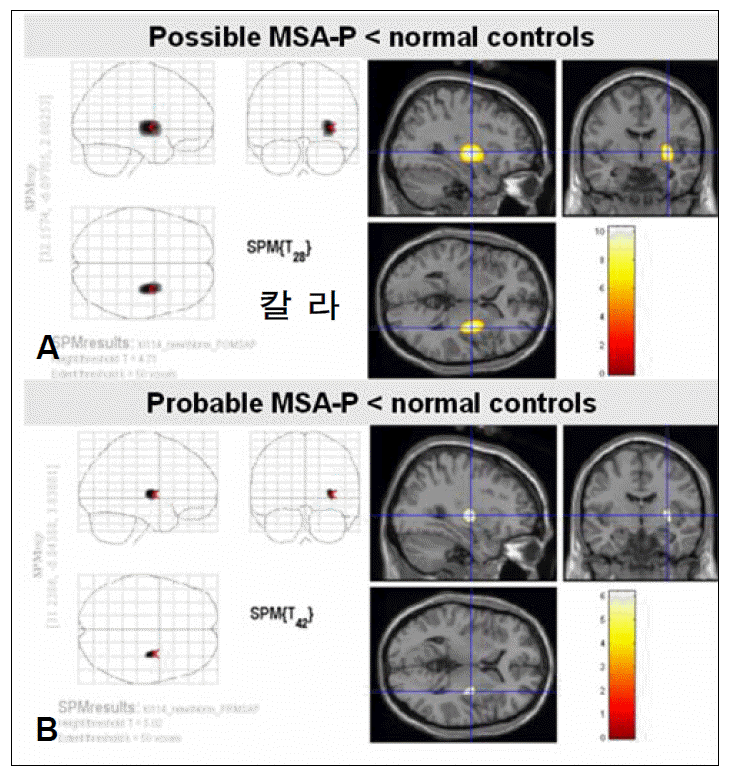
Figure 2.Comparison of putaminal metabolism between possible and probable Parkinsonian type multiple system atrophy (MSA-P) by 18F-FDG PET using the regions of interest (ROI) method. A greater decrease in glucose metabolism is revealed in the putamen contralateral to the body side with worse parkinsonism in patients with possible MSA-P than in those with probable MSA-P. Data are represented as median values (Mann-Whitney U test, p=0.018) and inter-quartile ranges.
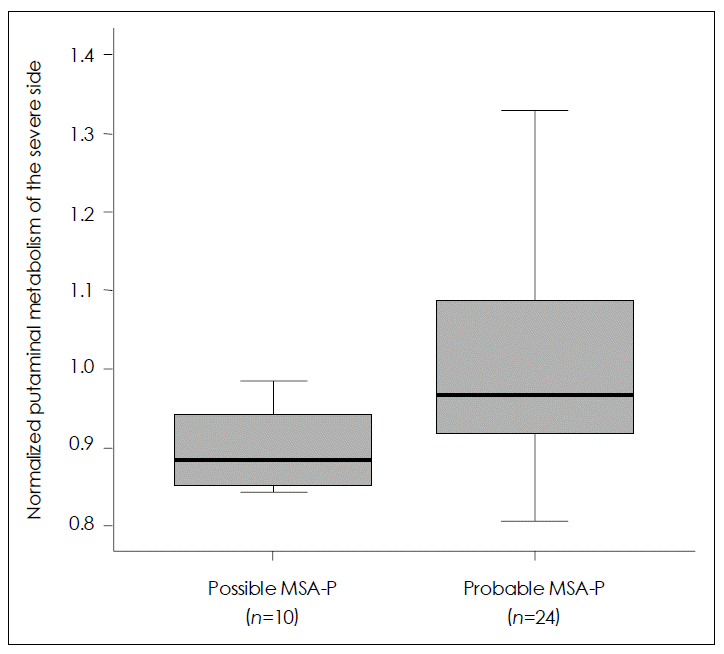
Figure 3.Statistical parametric mapping (SPM) analysis of possible and probable cerebellar type multiple system atrophy (MSAC) by 18F-FDG PET. A greater decrease in glucose metabolism is revealed in the cerebellum in patients with possible MSA-C (A) than in those with probable MSA-C (B) at a false discovery rate (FDR) corrected statistical threshold of p<0.01.

Figure 4.Comparison of cerebellar metabolism between possible and probable cerebellar type multiple system atrophy (MSA-C) by 18F-FDG PET using the regions of interest (ROI) method reveals a greater decrease in glucose metabolism in the cerebellum of patients with possible MSA-C than in those with probable MSA-C. Data are represented as median values (Mann-Whitney U test, p=0.018) and inter-quartile ranges.
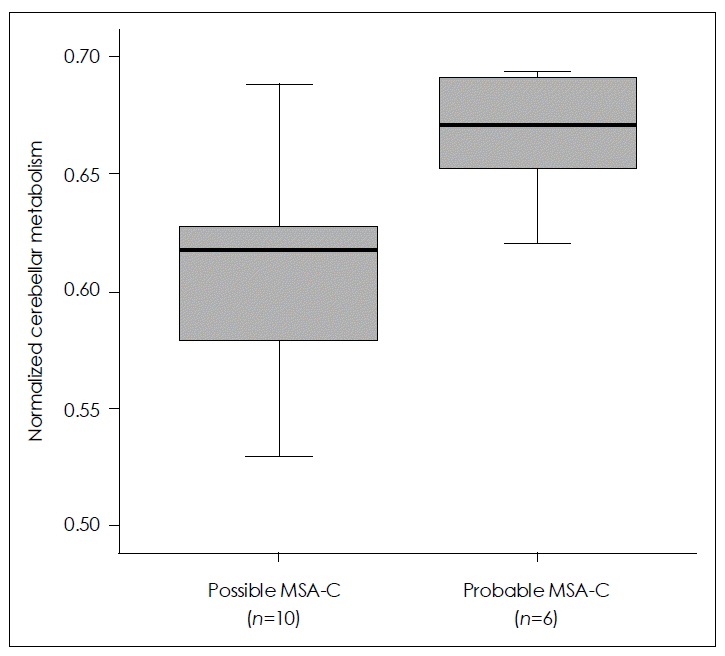
Table 1.Clinical characteristics of patients with MSA
Table 2.Clinical scales in the comparison between probable and possible MSA-P
- 1. Gilman S, Low P, Quinn N, Albanese A, Ben-Shlomo Y, Fowler C, et al. Consensus statement on the diagnosis of multiple system atrophy. American Autonomic Society and American Academy of Neurology. Clin Auton Res 1998;8:359–362.ArticlePubMed
- 2. Schrag A, Kingsley D, Phatouros C, Mathias CJ, Lees AJ, Daniel SE, et al. Clinical usefulness of magnetic resonance imaging in multiple system atrophy. J Neurol Neurosurg Psychiatry 1998;65:65–71.ArticlePubMedPMC
- 3. Schocke MF, Seppi K, Esterhammer R, Kremser C, Jaschke W, Poewe W, et al. Diffusion-weighted MRI differentiates the Parkinson variant of multiple system atrophy from PD. Neurology 2002;58:575–580.ArticlePubMed
- 4. Kraft E, Trenkwalder C, Auer DP. T2*-weighted MRI differentiates multiple system atrophy from Parkinson’s disease. Neurology 2002;59:1265–1267.ArticlePubMed
- 5. Minnerop M, Specht K, Ruhlmann J, Schimke N, Abele M, Weyer A, et al. Voxel-based morphometry and voxel-based relaxometry in multiple system atrophy-a comparison between clinical subtypes and correlations with clinical parameters. Neuroimage 2007;36:1086–1095.ArticlePubMed
- 6. Otsuka M, Kuwabara Y, Ichiya Y, Hosokawa S, Sasaki M, Yoshida T, et al. Differentiating between multiple system atrophy and Parkinson’s disease by positron emission tomography with 18F-dopa and 18F-FDG. Ann Nucl Med 1997;11:251–257.ArticlePubMed
- 7. Juh R, Pae CU, Lee CU, Yang D, Chung Y, Suh T, et al. Voxel based comparison of glucose metabolism in the differential diagnosis of the multiple system atrophy using statistical parametric mapping. Neurosci Res 2005;52:211–219.ArticlePubMed
- 8. Kwon KY, Choi CG, Kim JS, Lee MC, Chung SJ. Comparison of brain MRI and 18F-FDG PET in the differential diagnosis of multiple system atrophy from Parkinson’s disease. Mov Disord 2007;22:2352–2358.ArticlePubMed
- 9. Wenning GK, Ben Shlomo Y, Magalhães M, Daniel SE, Quinn NP. Clinical features and natural history of multiple system atrophy. An analysis of 100 cases. Brain 1994;117:835–845.ArticlePubMedPDF
- 10. Osaki Y, Wenning GK, Daniel SE, Hughes A, Lees AJ, Mathias CJ, et al. Do published criteria improve clinical diagnostic accuracy in multiple system atrophy? Neurology 2002;59:1486–1491.ArticlePubMed
- 11. Wenning GK, Tison F, Ben Shlomo Y, Daniel SE, Quinn NP. Multiple system atrophy: a review of 203 pathologically proven cases. Mov Disord 1997;12:133–147.ArticlePubMed
- 12. Ozawa T, Paviour D, Quinn NP, Josephs KA, Sangha H, Kilford L, et al. The spectrum of pathological involvement of the striatonigral and olivopontocerebellar systems in multiple system atrophy: clinicopathological correlations. Brain 2004;127:2657–2671.ArticlePubMedPDF
- 13. Jellinger KA, Seppi K, Wenning GK. Grading of neuropathology in multiple system atrophy: proposal for a novel scale. Mov Disord 2005;20(Suppl 12):S29–36.ArticlePubMed
- 14. Fahn S, Elton R. Unified Parkinson’s Disease Rating Scale. In: Fahn S, Marsden C, Calne D, Goldstein M, editors. Recent Developments in Parkinson’s Disease. Florham Park: Mac-Millan Health Care Information; 1987:153–163.
- 15. Wenning GK, Tison F, Seppi K, Sampaio C, Diem A, Yekhlef F, et al.; Multiple System Atrophy Study Group. Development and validation of the Unified Multiple System Atrophy Rating Scale (UMSARS). Mov Disord 2004;19:1391–1402.ArticlePubMed
- 16. Tzourio-Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N, et al. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. Neuroimage 2002;15:273–289.ArticlePubMed
- 17. Seppi K, Yekhlef F, Diem A, Luginger Wolf E, Mueller J, Tison F, et al. Progression of parkinsonism in multiple system atrophy. J Neurol 2005;252:91–96.ArticlePubMed
- 18. Geser F, Wenning GK, Seppi K, Stampfer-Kountchev M, Scherfler C, Sawires M, et al.; The European MSA Study Group. Progression of multiple system atrophy (MSA): a prospective natural history study by the European MSA Study Group (EMSA SG). Mov Disord 2006;21:179–186.ArticlePubMed
- 19. Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000;68:434–440.ArticlePubMedPMC
- 20. Watanabe H, Saito Y, Terao S, Ando T, Kachi T, Mukai E, et al. Progression and prognosis in multiple system atrophy: an analysis of 230 Japanese patients. Brain 2002;125:1070–1083.ArticlePubMedPDF
- 21. Juh R, Kim J, Moon D, Choe B, Suh T. Different metabolic patterns analysis of Parkinsonism on the 18F-FDG PET. Eur J Radiol 2004;51:223–233.ArticlePubMed
- 22. Eckert T, Barnes A, Dhawan V, Frucht S, Gordon MF, Feigin AS, et al. FDG PET in the differential diagnosis of parkinsonian disorders. Neuroimage 2005;26:912–921.ArticlePubMed
- 23. Taniwaki T, Nakagawa M, Yamada T, Yoshida T, Ohyagi Y, Sasaki M, et al. Cerebral metabolic changes in early multiple system atrophy: a PET study. J Neurol Sci 2002;200:79–84.ArticlePubMed
- 24. Antonini A, Leenders KL, Vontobel P, Maguire RP, Missimer J, Psylla M, et al. Complementary PET studies of striatal neuronal function in the differential diagnosis between multiple system atrophy and Parkinson’s disease. Brain 1997;120:2187–2195.ArticlePubMedPDF
- 25. Hughes AJ, Colosimo C, Kleedorfer B, Daniel SE, Lees AJ. The dopaminergic response in multiple system atrophy. J Neurol Neurosurg Psychiatry 1992;55:1009–1013.ArticlePubMedPMC
- 26. Colosimo C, Albanese A, Hughes AJ, de Bruin VM, Lees AJ. Some specific clinical features differentiate multiple system atrophy (striatonigral variety) from Parkinson’s disease. Arch Neurol 1995;52:294–298.ArticlePubMed
- 27. Schulz JB, Klockgether T, Petersen D, Jauch M, Müller-Schauenburg W, Spieker S, et al. Multiple system atrophy: natural history, MRI morphology, and dopamine receptor imaging with 123IBZM-SPECT. J Neurol Neurosurg Psychiatry 1994;57:1047–1056.ArticlePubMedPMC
- 28. Müller J, Wenning GK, Jellinger K, McKee A, Poewe W, Litvan I. Progression of Hoehn and Yahr stages in Parkinsonian disorders: a clinicopathologic study. Neurology 2000;55:888–891.ArticlePubMed
- 29. Rodriguez ME, Artieda J, Zubieta JL, Obeso JA. Reflex myoclonus in olivopontocerebellar atrophy. J Neurol Neurosurg Psychiatry 1994;57:316–319.ArticlePubMedPMC
- 30. Litvan I, Goetz CG, Jankovic J, Wenning GK, Booth V, Bartko JJ, et al. What is the accuracy of the clinical diagnosis of multiple system atrophy? A clinicopathologic study. Arch Neurol 1997;54:937–944.ArticlePubMed
- 31. Hughes AJ, Daniel SE, Ben-Shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain 2002;125:861–870.ArticlePubMedPDF
- 32. Ito H, Kusaka H, Matsumoto S, Imai T. Striatal efferent involvement and its correlation to levodopa efficacy in patients with multiple system atrophy. Neurology 1996;47:1291–1299.ArticlePubMed
REFERENCES
Figure & Data
References
Citations
Citations to this article as recorded by 
- F-18 FP-CIT PET in Multiple System Atrophy of the Cerebellar Type: Additional Role in Treatment
Young Jin Jeong, Sang-Myung Cheon, Do-Young Kang, Jae Woo Kim
Contrast Media & Molecular Imaging.2017; 2017: 1. CrossRef - A systematic review of lessons learned from PET molecular imaging research in atypical parkinsonism
Flavia Niccolini, Marios Politis
European Journal of Nuclear Medicine and Molecular Imaging.2016; 43(12): 2244. CrossRef
Comments on this article
 PubReader
PubReader ePub Link
ePub Link Cite
Cite